 PDF
PDF  Views
Views  Share
Share


The Rare Face of an Infrequent Lymphoma Diagnosed by an Exquisite Method: Case Report with Review of Literature
CC BY-NC-ND 4.0 ? Indian J Med Paediatr Oncol 2021; 42(05): 501-505
DOI: DOI: 10.1055/s-0041-1735518
Abstract
Large B-cell lymphomas include both the commonly encountered, as well as not so frequent entities. We report a rare case of hepatic intravascular large B-cell lymphoma (IVLBCL). These cases usually pose a major challenge to timely diagnosis due to the limited clinical exposure and nonspecific symptoms; however, timely diagnosis is possible with awareness of this entity and close coordination between radiology and pathology. The patient presented with pain abdomen, fever, edema, hepatomegaly but no lymphadenopathy or cutaneous lesions were identified. Blood investigations revealed pancytopenia, high lactate dehydrogenase (LDH) levels and bone marrow investigations were not helpful. Left-sided portal hypertension, coagulopathy, and hypoalbuminemia raised a clinical suspicion of chronic liver disease with high LDH levels and liver biopsy was challenging in view of very low platelet counts. The uniquely explored transjugular biopsy helped in obtaining liver tissue which showed sinusoidal large atypical lymphoid cells positive for CD20 rendering a diagnosis of IVLBCL. After administration of the R-miniCHOP chemotherapy, there was evident clinical and radiological response. This case highlights the importance of considering transjugular liver biopsy in scenarios where the clinical presentation is unusual. At the same time, a meticulous histological examination is needed to diagnose a rare yet potentially treatable large B cell lymphoma.
Ethical Consideration
Informed consent was obtained from the patient to use the material and case details for academic purposes.
Author Contributions
D.D. diagnosed the histology case. M.P. interpreted the cytogenetics report. M.P., DD, and B.S.L. prepared the manuscripts. S.J.B. and S.C. were responsible for treating the patient. D.L. did the transjugular liver biopsy, while J.D interpreted the pre and postchemotherapy positron emission tomography -computed tomography (PET-CT) scans. All authors read and approved the manuscripts.
Supplementary Material
Publication History
Publication Date:
24 December 2021 (online)
? 2021. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Large B-cell lymphomas include both the commonly encountered, as well as not so frequent entities. We report a rare case of hepatic intravascular large B-cell lymphoma (IVLBCL). These cases usually pose a major challenge to timely diagnosis due to the limited clinical exposure and nonspecific symptoms; however, timely diagnosis is possible with awareness of this entity and close coordination between radiology and pathology. The patient presented with pain abdomen, fever, edema, hepatomegaly but no lymphadenopathy or cutaneous lesions were identified. Blood investigations revealed pancytopenia, high lactate dehydrogenase (LDH) levels and bone marrow investigations were not helpful. Left-sided portal hypertension, coagulopathy, and hypoalbuminemia raised a clinical suspicion of chronic liver disease with high LDH levels and liver biopsy was challenging in view of very low platelet counts. The uniquely explored transjugular biopsy helped in obtaining liver tissue which showed sinusoidal large atypical lymphoid cells positive for CD20 rendering a diagnosis of IVLBCL. After administration of the R-miniCHOP chemotherapy, there was evident clinical and radiological response. This case highlights the importance of considering transjugular liver biopsy in scenarios where the clinical presentation is unusual. At the same time, a meticulous histological examination is needed to diagnose a rare yet potentially treatable large B cell lymphoma.
Introduction
World Health Organization (WHO) 2016 classification lists large B-cell lymphomas as different common and rare subtypes. While the frequent ones are readily diagnosed, the rare entities pose clinical and pathological challenges with respect to early diagnosis and treatment.[1] Intravascular large B-cell lymphoma (IVLBCL) is one such extremely rare subtype of large B-cell lymphoma with reported incidence of <1 href="https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0041-1735518#JR211440217-2" xss=removed>2] It was first described by Lorenz Pfleger and Jasmin Tappeiner in 1959 as vascular endothelial neoplasm, namely, ?angioendotheliomatosis proliferans systemisata.?[3] Its rarity, unique presentation in extranodal organs, and unsuspected clinical presentation pose a challenge to timely diagnosis and management.[4] This is compounded by the fact that there is no definite organ to biopsy. Therefore, most patients were diagnosed at autopsy in the past.[5] However, a high index of clinical suspicion and targeting sites like skin, bone marrow, and liver for the biopsy help in early diagnosis.[5]
We report such a diagnostically challenging case of hepatic IVLBCL which presented as hepatosplenomegaly, coagulopathy, hypoalbuminemia, and pancytopenia.
Case Report
A 74-year-old woman of Oriental origin with no associated known comorbidities presented with fever, pain in abdomen, loss of appetite, and dyspepsia of 5 months. She consulted a physician at a local hospital in her hometown wherein the investigation included endoscopy to ascertain the cause of dyspepsia and abdomen pain. Endoscopy revealed pangastritis with associated?Helicobacter pylori?infection detected on positive rapid urease test for which she was administered triple therapy regimen. However, she developed adverse reaction to clarithromycin as generalized body rash. The triple therapy was stopped, and she was given prednisolone and omeprazole. The probable rationale of therapy was to treat the body rash and gastritis, respectively. While the patient was on this treatment, pain in abdomen recurred along with right lower limb edema, diarrhea, fever, hypotension, thrombocytopenia, and leucopenia. She was transferred to our medical facility. At the time of presentation, the patient had fever and without any history of weight loss or sweating. On palpation, massive hepatosplenomegaly was found. The blood investigations showed severe pancytopenia (hemoglobin, 7.5?g/dL; TLC (total leucocyte count), 1,000/?L, and platelet count, 25,000/?L), coagulopathy (APTT [activated partial thromboplastin time] >245?seconds; PT [prothrombin time] with international normalized ratio [INR] of 12.4 second; and fibrinogen, 139?mg/dL), raised ferritin (213?ng/mL), and vitamin B12 (>2,000 pg/mL) with hypoalbuminemia (1.4?g/dL), raised serum glutamic-oxaloacetic transaminase (SGOT) of 68 U/L, and total protein of 3?g/dL. Her lactate dehydrogenase (LDH) levels were markedly elevated at 1,200 U/L, and erythrocyte sedimentation rate (ESR) was 90?mm/first hour. Lymphadenopathy or cutaneous lesions were not found on physical examination. Therefore, lymph node biopsy or skin biopsy was not thought of.
Endoscopy was planned for complaints of dyspepsia and pain abdomen to ascertain the etiology. It showed minimal portal venous gastropathy but no varices. Doppler study revealed dilated portal vein (likely sinusoidal cause), massive splenomegaly, and moderate ascites. Echocardiography detected moderate pulmonary artery hypertension with grade-1 diastolic dysfunction. Left ventricular cavity size was normal with ejection fraction of 60%.
Computed tomography (CT) scan reported hepatosplenomegaly (liver, 17.3?cm and spleen, 16.8?cm), splenic infarct, dilated portal vein with splenic collaterals, ascites, and bilateral pleural effusion. Positron emission tomography?computed tomography (PET-CT) scan highlighted diffuse metabolic uptake in the enlarged spleen, avid bilateral pleural effusion, and diffuse subcutaneous edema ([Fig. 1A]). Bone marrow examination showed cellular aspirate with trilineage hematopoiesis without any abnormal cells. There was an increase in monohistiocytic cells with few showing hemophagocytosis. CD68 done on bone marrow biopsy highlighted the mono-histiocytic cells. Since there was no clinical suspicion of lymphoma and the marrow was reactive on morphology without any suspicious or abnormal cells, flow cytometry was not done.
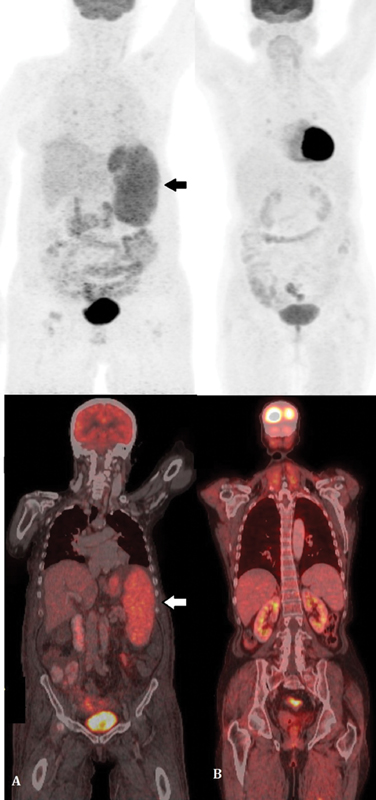
|?Fig. 1? Positron emission tomography?computed tomography (PET-CT) scan of the patient. (A) The pre therapy scan shows diffuse metabolic uptake in the enlarged spleen, (B) posttherapy scan revealed no metabolically active disease focus, thus indicating complete response. Arrow depicts ?large atypical lymphoid cells?.|
Cytogenetic analysis showed the presence of a complex karyotype {Karotyping:41?46, XX,del(3)(q21),6,7,8,der(10)t(10;?)(q25;?),del(13)(q12q32),der(16)t(16;?)(q12?q13;?), der(18)t(18;?)(p11.1;?),der(19)t(19;?)(p13;?),?+?2?7mar[cp5]/46,XX[15]}([Supplementary Fig. S1]; available in the online version).
The presence of left-sided portal hypertension, coagulopathy, and hypoalbuminemia raised a clinical suspicion of chronic liver disease with high LDH levels and hence liver biopsy was planned. The percutaneous liver biopsy was contraindicated due to pancytopenia and hence transjugular biopsy was performed. Hepatic vein-portal-vein pressure gradient (HVPG) measured during the procedure was within normal limits. Although lymphoma was not suspected clinically at the time of biopsy but surprisingly, the transjugular liver biopsy displayed few large, atypical lymphoid cells within the sinusoids. They exhibited pleomorphic nuclei along with mitotic figures and apoptotic bodies ([Fig. 2A] and [B]). The overall hepatic architecture was preserved with background macrovesicular and microvesicular steatosis and nuclear glycogenation. No bridging fibrosis or cirrhosis was seen. Immunohistochemistry found the large sinusoidal lymphoid cells to be positive for CD20, bcl6 (weak), bcl2 and MUM-1 ([Fig. 2C?E]), while they were negative for CD30, TdT, CD138 and Cyclin-D1. The c-myc stained 40% of the neoplastic B-cell population while Ki-67 proliferation index was around 70% in these cells. A diagnosis of IVLBCL without cirrhosis was rendered. The patient was treated with six cycles of R-miniCHOP which comprised rituximab, 375?mg/m2; cyclophosphamide, 400?mg/m2; adriamycin, 25?mg/m2; vincristine, 1?mg/m2; and prednisolone, 30?mg/m2?daily once for 5 days. She responded with increase in blood counts and albumin with subsequent decrease in pedal edema. At 6-month follow-up, the patient was doing well with normal blood counts and minimal edema. On repeat PET-CT scan, no metabolically active disease focus was detected indicating complete response ([Fig. 1B]).
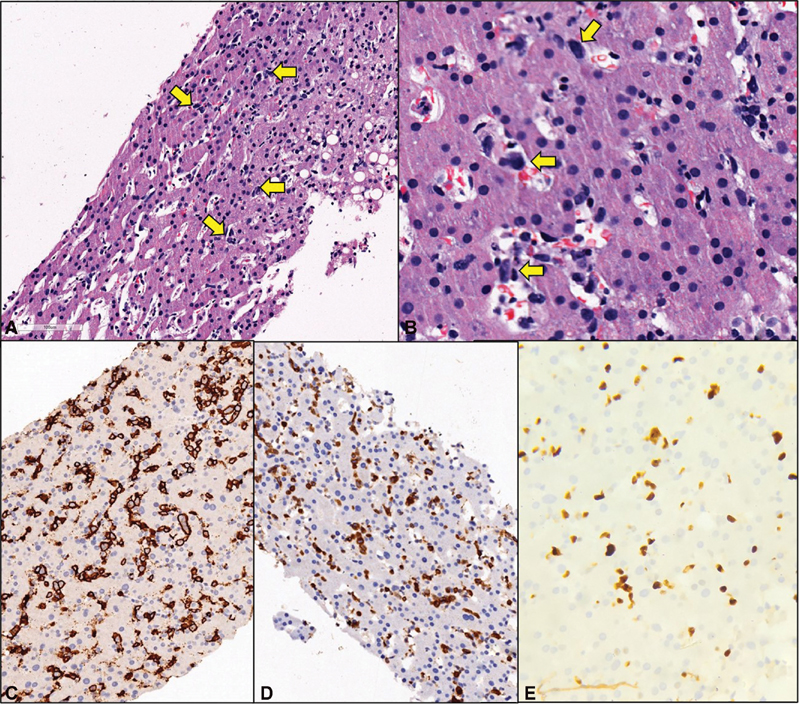
|?Fig. 1? Histopathological and immunohistochemical results. (A) The liver biopsy displayed few large, atypical lymphoid cells within the sinusoids (H&E, ?10). (B) These cells exhibited pleomorphic nuclei along with mitotic figures and apoptotic bodies (H&E, ?40). Immunohistochemistry found the large sinusoidal lymphoid cells to be positive for CD20 (C), bcl2 (D), while they were negative for CD30, TdT, CD138 and Cyclin-D1 (not included). The Ki-67 proliferation index within the CD20 positive population was around 70%. (E). H&E, hematoxylin and eosin.|
Discussion
In 2008, WHO classification described IVLBCL as a lymphoma characterized by the presence of neoplastic cells within the lumen of small blood vessels.[1] IVLBCL cells are devoid of molecules like CD29 (?1 integrin subunit) which are important for lymphocytes extravasation. Additionally, homeostatic chemokine receptors, CXCR5, CCR6, and CCR7 are decreased in this lymphoma which act on lymphocyte migration across the structures.[5] Thus these lymphoma cells occur within the lumen of blood vessels, and it is critical in distinguishing IVLBCL from diffuse large B-cell lymphomas (DLBCL).
This rare lymphoma predominantly affects adults in sixth and seventh decades of life with median age of 67 years and no gender predilection.[1] [2] The clinical presentation characteristically shows geographic differentiation between the western and eastern population. The ?classic? form of disease is witnessed in Caucasian patients while the ?hemophagocytic syndrome associated (HAS)? form occurs in Asian patients. Fever invariably occurs in 55 to 76% of patients of both the forms.[1] It is the most common presenting symptom in the classic form, even more frequent (45%) than other aggressive extranodal lymphomas (25%). Cutaneous and neurologic symptoms at diagnosis are reported in 40 and 35% of patients with classic form of the disease, respectively. Lungs involvement and endocrine organs involvement (mainly thyroid, adrenal, and pituitary glands) may be encountered in the classic variant.[2]
On the contrary, HAS variant typically presents as hemophagocytic syndrome with bone marrow involvement, fever, hepatosplenomegaly, and thrombocytopenia occurring in 73 to 100% patients.[5] [6] An exclusive cutaneous variant, occurring in females, is recognized by normal leukocyte and platelet counts, monoclonal component is rarely observed, Eastern Cooperative Oncology Group-performance status (ECOG-PS) is usually ?1 and the disease is comparatively less aggressive than the other two variants.[1] [5] Due to different clinical presentation in all variants, the latest WHO classification has suggested to classify them based on clinical features, into classic form, cutaneous, and hemophagocytic syndrome associated, rather than by geographical distribution.[5]
Close to 15% of the IVLBCL cases are associated with other benign nonhematological neoplasms, including vascular tumors, soft tissue tumors, and epithelial carcinomas. In such cases, the IVLBCL cells are found within the blood vessels of the tumor raising the possibility of distinct molecular expression by these cancer associated endothelia.[5] [7]
Laboratory findings in both the former variants include leucopenia or thrombocytopenia associated with anemia (in 63% patients), hypoalbuminemia (almost in 100% patients), elevated ESR (in 43% patients), and extremely elevated LDH levels.[1] [5] [6] [7] Our patient was an elderly female of Oriental origin and had fever, splenomegaly, pancytopenia with neutropenia, elevated ferritin level, hypofibrinogenemia, hemophagocytosis in bone marrow, all of which are a constituent of hemophagolymphohistiocytosis (HLH). In addition, splenomegaly may have contributed to pancytopenia.
In a large series of IVLBCL, most patients (69%) were diagnosed with random skin biopsies followed by bone marrow biopsy alone (19%).[6] Bone marrow biopsy, in our case did not show any morphological abnormality; however, hemophagocytosis was detected and cytogenetics revealed a complex karyotype. The transjugular liver biopsy, which clinched the diagnosis in our case, was initially considered as an inferior specimen, but recent studies report a successful histological diagnosis in 95% biopsies performed.[8]
The neoplastic cells in IVLBCL express mature B-cell immunophenotype wherein CD20 expression is almost a rule which also aids in effective treatment with rituximab. In exceptional cases where CD20 is negative, alternate B-cell markers like PAX-5 and/or CD79a help in establishing the diagnosis. In our case, the large sinusoidal lymphoid cells were positive for CD20, bcl6 (weak), bcl2, and MUM-1 ([Fig. 2C?E]), while they were negative for CD30, TdT, CD138, and Cyclin-D1.
IVLBCL does not have specific chromosomal alterations. The most common of such patterns include -6 or 6q- and dup(18q) or +18, with commonly amplified region located at 18q13-q23 and minimally deleted region located at 6q21-q23.[3]
Fully reliable staging parameters for IVLBCL are not available. In around 40% of patients, who are incidentally diagnosed or who display the cutaneous form of the disease, stage IE?by Ann Arbor staging system is rendered. The rest 60% patients usually present with stage-IV disease. Staging workup therefore includes magnetic resonance imaging and bone marrow biopsy. Hepatic, thyroid, or renal function tests may be used as surrogates of organs involvement for staging.[5] With the exception of cutaneous variant, IVLBCL is an aggressive disseminated disease and treated likewise. Our patient showed involvement of liver and hence was in stage IV at presentation.
The outcomes of IVLBCL were dismal in the prerituximab era when steroid therapy gave tentative results. Shimada et al found 2 years of progression-free survival and overall survival as 56 and 66%, respectively, in patients receiving chemotherapy with rituximab, whereas it was 27 and 46%, respectively, in those not receiving rituximab.[7] The R-CHOP therapy is mainly used for treatment in clinical practice and few authors have reported complete response.[9] The elevated complement concentrations and higher drug bioavailability in the lumina of small vessels explains this positive effect of rituximab. Central nervous system (CNS) prophylaxis and treatment is equally important since the extravascular CNS dissemination of the disease accounts for most relapsing IVLBCL patients treated with R-CHOP. The possible strategy is to add drugs with better CNS bioavailability like high-dose methotrexate.[5]
In a retrospective study, autologous stem-cell transplantation showed clinical improvement as compared with R-CHOP therapy. However, number of eligible patients are very few since IVLBCL occurs in seventh decade of life and generally patients have poor PS.[5] Our patient also showed good response to the therapy as evident by laboratory investigations and PET-CT scan.
Conclusion
We highlight that a high index of suspicion is needed to detect IVLBCL even when the routine bone marrow biopsy does not help. The clinical and interventional radiology team must explore alternate options to obtaining biopsy-like transjugular liver biopsy in this case. At the same time, presence of neoplastic cells only in the sinusoids can easily be missed on a cursory glance and hence detailed analysis by pathologist is utmost important. IVLBCL was initially considered fatal but now with R-CHOP therapy, it is amenable to treatment.
Conflicts of Interest
None declared.
Acknowledgment
The authors would like to acknowledge Dr. Deepak K. Mishra for the bone marrow report and Dr. Lateef Zameer for his inputs.
Ethical Consideration
Informed consent was obtained from the patient to use the material and case details for academic purposes.
Author Contributions
D.D. diagnosed the histology case. M.P. interpreted the cytogenetics report. M.P., DD, and B.S.L. prepared the manuscripts. S.J.B. and S.C. were responsible for treating the patient. D.L. did the transjugular liver biopsy, while J.D interpreted the pre and postchemotherapy positron emission tomography -computed tomography (PET-CT) scans. All authors read and approved the manuscripts.
Supplementary Material
References
- Nakamura S, Ponzoni M, Campo E.?Intravascular large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. eds. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. 4th ed.. Lyon, France: International Agency for Research on Cancer; 2017: 317-318
- Rajyaguru DJ, Bhaskar C, Borgert AJ, Smith A, Parsons B.?Intravascular large B-cell lymphoma in the United States (US): a population-based study using Surveillance, Epidemiology, and End Results program and National Cancer Database. Leuk Lymphoma 2017; 58 (09) 1-9
- Orwat DE, Batalis NI.?Intravascular large B-cell lymphoma. Arch Pathol Lab Med 2012; 136 (03) 333-338
- Ramkumar B, Hutchison R, Khadim H, Gajra A.?Intravascular large B-cell lymphoma?a diagnostic dilemma. Clin Lymphoma Myeloma Leuk 2013; 13 (06) e18-e21
- Ponzoni M, Campo E, Nakamura S.?Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood 2018; 132 (15) 1561-1567
- Matsue K, Abe Y, Narita K. et al.?Diagnosis of intravascular large B cell lymphoma: novel insights into clinicopathological features from 42 patients at a single institution over 20 years. Br J Haematol 2019; 187 (03) 328-336
- Shimada K, Kinoshita T, Naoe T, Nakamura S.?Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol 2009; 10 (09) 895-902
- Stift J, Semmler G, Walzel C. et al.?Transjugular aspiration liver biopsy performed by hepatologists trained in HVPG measurements is safe and provides important diagnostic information. Dig Liver Dis 2019; 51 (08) 1144-1151
- Sekiguchi Y, Shimada A, Imai H. et al.?Intravascular large B-cell lymphoma with pontine involvement successfully treated with R-CHOP therapy and intrathecal administration: a case report and review of literature. Int J Clin Exp Pathol 2014; 7 (06) 3363-3369
Address for correspondence
Publication History
Publication Date:
24 December 2021 (online)
? 2021. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
|?Fig. 1? Positron emission tomography?computed tomography (PET-CT) scan of the patient. (A) The pre therapy scan shows diffuse metabolic uptake in the enlarged spleen, (B) posttherapy scan revealed no metabolically active disease focus, thus indicating complete response. Arrow depicts ?large atypical lymphoid cells?.|
|?Fig. 1? Histopathological and immunohistochemical results. (A) The liver biopsy displayed few large, atypical lymphoid cells within the sinusoids (H&E, ?10). (B) These cells exhibited pleomorphic nuclei along with mitotic figures and apoptotic bodies (H&E, ?40). Immunohistochemistry found the large sinusoidal lymphoid cells to be positive for CD20 (C), bcl2 (D), while they were negative for CD30, TdT, CD138 and Cyclin-D1 (not included). The Ki-67 proliferation index within the CD20 positive population was around 70%. (E). H&E, hematoxylin and eosin.|
References
- Nakamura S, Ponzoni M, Campo E.?Intravascular large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. eds. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. 4th ed.. Lyon, France: International Agency for Research on Cancer; 2017: 317-318
- Rajyaguru DJ, Bhaskar C, Borgert AJ, Smith A, Parsons B.?Intravascular large B-cell lymphoma in the United States (US): a population-based study using Surveillance, Epidemiology, and End Results program and National Cancer Database. Leuk Lymphoma 2017; 58 (09) 1-9
- Orwat DE, Batalis NI.?Intravascular large B-cell lymphoma. Arch Pathol Lab Med 2012; 136 (03) 333-338
- Ramkumar B, Hutchison R, Khadim H, Gajra A.?Intravascular large B-cell lymphoma?a diagnostic dilemma. Clin Lymphoma Myeloma Leuk 2013; 13 (06) e18-e21
- Ponzoni M, Campo E, Nakamura S.?Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood 2018; 132 (15) 1561-1567
- Matsue K, Abe Y, Narita K. et al.?Diagnosis of intravascular large B cell lymphoma: novel insights into clinicopathological features from 42 patients at a single institution over 20 years. Br J Haematol 2019; 187 (03) 328-336
- Shimada K, Kinoshita T, Naoe T, Nakamura S.?Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol 2009; 10 (09) 895-902
- Stift J, Semmler G, Walzel C. et al.?Transjugular aspiration liver biopsy performed by hepatologists trained in HVPG measurements is safe and provides important diagnostic information. Dig Liver Dis 2019; 51 (08) 1144-1151
- Sekiguchi Y, Shimada A, Imai H. et al.?Intravascular large B-cell lymphoma with pontine involvement successfully treated with R-CHOP therapy and intrathecal administration: a case report and review of literature. Int J Clin Exp Pathol 2014; 7 (06) 3363-3369