 PDF
PDF  Views
Views  Share
Share


Structure-Based Virtual Screening and Protein–Protein Docking Analysis of ERBB2 and Associated Proteins for Pediatric Cancer Therapeutic Approaches
CC BY 4.0 · Indian J Med Paediatr Oncol 2025; 46(01): 064-070
DOI: 10.1055/s-0044-1786163
Abstract
Introduction The Erythroblastosis Oncogene B homolog 2 (ERBB2) protein, also known as human epidermal growth factor receptor 2 (HER2), is a key player in cancer growth, especially in neuroblastoma and gastric cancers. Targeting ERBB2 has led to successful therapies, making it an important focus in cancer research with the potential to improve treatment for HER2-positive cancers.
Objective The primary goal of this research is to employ a multifaceted computational approach to identify potential drug candidates targeting ERBB2. We aim to combine virtual screening, protein–protein docking, and functional partner prediction to provide insights into the molecular interactions and potential efficacy of the identified compounds. Additionally, we intend to assess the safety profiles of these compounds using advanced toxicity prediction tools.
Materials and Methods Relevant protein sequence and structural data for ERBB2 and epidermal growth factor receptor (EGFR) were sourced from publicly available databases. Potential inhibitors from the Enamine and LifeChemicals databases were identified through virtual screening using AutoDock Vina. Functional partners of ERBB2 were explored using STRING, KEGG, and REACTOME servers. The identified compounds were subjected to toxicity prediction using the ProTox-II server.
Results Virtual screening led to the selection of 10 compounds with favorable binding energies (–8.346 to –6.296 kcal/mol) and specific amino acid interactions (Thr5, Arg412, Leu414, and Ser441) with the receptor. On the other hand, EGFR was identified as the best functional partner for ERBB2. The EGFR residues Gln408, Lys463, Phe412, and Asp436 found key residues for the complex formation. The toxicity prediction analysis revealed that the majority of compounds exhibited acceptable safety profiles, although a subset of compounds showed lower prediction scores, suggesting the need for further consideration.
Conclusion This comprehensive computational approach, integrating virtual screening, protein–protein docking, functional partner identification, and toxicity prediction, offers a systematic framework for efficient drug discovery. The identification of potential lead compounds targeting ERBB2, with emphasis on both binding affinity and safety, underscores the significance of such an approach in streamlining the drug development process. By prioritizing compounds with promising efficacy, functional relevance, and acceptable toxicity profiles, this study advances our understanding of potential therapeutic agents, enhancing the likelihood of successful translation from computational predictions to real-world drug candidates.
Publication History
Article published online:
02 September 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
- Discovery of Tyrosinase Inhibitors: Structure-Based Virtual Screening and Biological EvaluationPharmaceutical Fronts, 2022
- Search for cannabinoid receptor 1 antagonists using structure-based virtual screening: identification of natural product hitsP Pandey, Planta Medica, 2014
- Search for cannabinoid receptor 1 antagonists using structure-based virtual screening: identification of natural product hitsP Pandey, Planta Medica, 2014
- Neurologic Manifestations of Pediatric Acquired Immunodeficiency Syndrome: Clinical Features and Therapeutic ApproachesMark Mintz, Seminars in Neurology, 1992
- Neurologic Manifestations of Pediatric Acquired Immunodeficiency Syndrome: Clinical Features and Therapeutic ApproachesMark Mintz, Seminars in Neurology, 1992
- Materials-Based Approaches for Cancer VaccinationMiguel C. Sobral, J Immunol, 2024
- Cancer-associated fibroblasts: heterogeneity, tumorigenicity and therapeutic targetsKangzheng Lv, et al., Molecular Biomedicine, 2024
- STAT reporter cell line systems as a tool for cancer therapeutic target screening.Hyun-Ku Lee, J Immunol, 2018
- Design, synthesis and biological evaluation of novel histone deacetylase inhibitors based on virtual screeningHui Lu, Acta Pharmaceutica Sinica B
- The Diagnostic Accuracy of an Electronic Medical Record-Based Screening Tool for Pediatric Pneumonia.Nancy S. Rixe, Pediatrics, 2021
Abstract
Introduction The Erythroblastosis Oncogene B homolog 2 (ERBB2) protein, also known as human epidermal growth factor receptor 2 (HER2), is a key player in cancer growth, especially in neuroblastoma and gastric cancers. Targeting ERBB2 has led to successful therapies, making it an important focus in cancer research with the potential to improve treatment for HER2-positive cancers.
Objective The primary goal of this research is to employ a multifaceted computational approach to identify potential drug candidates targeting ERBB2. We aim to combine virtual screening, protein–protein docking, and functional partner prediction to provide insights into the molecular interactions and potential efficacy of the identified compounds. Additionally, we intend to assess the safety profiles of these compounds using advanced toxicity prediction tools.
Materials and Methods Relevant protein sequence and structural data for ERBB2 and epidermal growth factor receptor (EGFR) were sourced from publicly available databases. Potential inhibitors from the Enamine and LifeChemicals databases were identified through virtual screening using AutoDock Vina. Functional partners of ERBB2 were explored using STRING, KEGG, and REACTOME servers. The identified compounds were subjected to toxicity prediction using the ProTox-II server.
Results Virtual screening led to the selection of 10 compounds with favorable binding energies (–8.346 to –6.296 kcal/mol) and specific amino acid interactions (Thr5, Arg412, Leu414, and Ser441) with the receptor. On the other hand, EGFR was identified as the best functional partner for ERBB2. The EGFR residues Gln408, Lys463, Phe412, and Asp436 found key residues for the complex formation. The toxicity prediction analysis revealed that the majority of compounds exhibited acceptable safety profiles, although a subset of compounds showed lower prediction scores, suggesting the need for further consideration.
Conclusion This comprehensive computational approach, integrating virtual screening, protein–protein docking, functional partner identification, and toxicity prediction, offers a systematic framework for efficient drug discovery. The identification of potential lead compounds targeting ERBB2, with emphasis on both binding affinity and safety, underscores the significance of such an approach in streamlining the drug development process. By prioritizing compounds with promising efficacy, functional relevance, and acceptable toxicity profiles, this study advances our understanding of potential therapeutic agents, enhancing the likelihood of successful translation from computational predictions to real-world drug candidates.
Keywords
AutoDock Vina - docking - ERBB2 - EGFR - toxicity prediction - virtual screeningIntroduction
The ERBB2 protein, also known as human epidermal growth factor receptor 2 (HER2), plays a pivotal role in cell proliferation, survival, and differentiation.[1] Dysregulation or overexpression of ERBB2 has been linked to various cancers, such as neuroblastoma, gastric, breast, and ovarian cancers, making it an attractive therapeutic target.[2] Human ERBB2 was initially identified as an oncogene in rat brain tumors induced by chemicals. Subsequent analysis of human tissues revealed ERBB2 amplification in specific cases of salivary carcinomas and breast cancers with poor prognosis. These early findings sparked significant interest in ERBB2's role in human cancer, leading to a multitude of studies investigating the biology and clinical relevance of ERBB receptor signaling.[3] [4] In recent years, structure-based virtual screening and in silico analysis have emerged as powerful approaches to identifying potential inhibitors for specific protein targets.
This comprehensive study aimed to identify novel inhibitors for the ERBB2 protein through structure-based virtual screening and in silico analysis. Leveraging the wealth of available protein structural data, computational tools, and advanced algorithms, we sought to identify small molecules with the potential to interact with key binding sites on ERBB2 and disrupt its activity.
By employing state-of-the-art computational techniques, including molecular docking, protein–protein docking, and binding free energy calculations, we conducted an extensive screening of a diverse chemical library. Our focus on ERBB2-specific binding sites aims to prioritize compounds with high binding affinities and favorable amino acid interactions, thereby increasing the likelihood of successful inhibition. The identification of novel ERBB2 inhibitors holds promise for developing targeted therapies that can effectively combat cancers associated with ERBB2 dysregulation.[5] These findings may contribute significantly to advancing personalized medicine and improving the overall efficacy of cancer treatments.
Overall, this study represents a crucial step toward harnessing the power of computational approaches to expedite the discovery of new and potent ERBB2 inhibitors, fostering advancements in precision oncology and targeted therapeutics. The implications of these findings in the context of cancer therapy and future directions for experimental validation and drug development are also discussed.
Methodology
Data Collection
Relevant protein sequence and structural data for ERBB2 and other targets were sourced from publicly available databases and repositories, including PubMed, RCSB-PDB, and UniProt.[6] Our primary objective was to identify potential inhibitors from diverse natural databases, such as Enamine and LifeChemicals for ERBB2.[7] [8] These databases are distinguished for their wealth of natural compound derivatives and medicinal value. The compounds pinpointed through virtual screening present viable candidates for subsequent experimental studies. The compound library was downloaded from the official website of the respective chemical compound database. A protein–protein docking protocol and pertinent data were acquired through a comprehensive literature survey.
Protein and Ligand Preparation
Protein and ligand preparation are essential steps in molecular docking studies, and AutoDock Vina serves as a powerful tool for this purpose. In the initial phase, the protein structure is prepared by removing any water molecules, heteroatoms, and cocrystallized ligands that are not part of the binding site. The protein is then assigned appropriate atom types, charges, and torsion angles, and polar hydrogens are added.[9] Careful attention is given to the correct protonation states of ionizable residues, ensuring accuracy in the simulation.
On the other hand, ligands are prepared by removing any counterions, water molecules, or other nonessential entities. The ligand's three-dimensional structure is refined by optimizing bond lengths, angles, and torsion angles. Proper charges, atom types, and hybridization states are assigned to the ligand's atoms, ensuring compatibility with the chosen force field. Additionally, for flexible ligand docking, multiple conformations of the ligand were generated for a single ligand to explore potential binding modes. Based on the binding energy calculated for each ligand conformation, the potential ligand and its binding pose were considered.
Overall, this meticulous preparation of both protein and ligand ensures a reliable and accurate docking simulation with AutoDock Vina, enabling the exploration of protein–ligand interactions and the prediction of potential binding poses, ultimately aiding in drug discovery and future molecular design efforts.
Structure-Based Virtual Screening
Structure-based virtual screening is a valuable computational approach employed to identify potential drug candidates by predicting their binding affinities to a target protein (ERBB2), using the AutoDock Vina tool.[10] In this study, the LifeChemicals and Enamine compound databases were utilized as valuable sources of diverse small molecules. The prepared protein structure and ligand molecules were utilized for the virtual screening studies.
The virtual screening was conducted by docking each compound from the LifeChemicals and Enamine databases into the active site of the ERBB2 protein. AutoDock Vina exhaustively sampled binding poses and ranked the compounds based on their calculated binding energies, reflecting the strength of their potential interactions with the protein. The top-ranking compounds, with the most favorable binding energies, were further analyzed to assess their predicted binding modes, hydrogen bonding patterns, and key interacting residues within the binding site. Default protocols of the AutoDock Vina were implemented throughout the virtual screening analysis.
This structure-based virtual screening using AutoDock Vina, coupled with the utilization of the LifeChemicals and Enamine databases, provided a systematic and efficient means to prioritize promising small molecules for potential ERBB2 inhibition.[11] [12] The results from this study contribute valuable insights into the realm of drug discovery, guiding experimental efforts toward the identification and development of novel therapeutic agents targeting ERBB2-associated diseases.[13]
Functional Partner Discovery with Bioinformatics Tools
Functional partners in a pathway are crucial components that interact with each other to execute specific biological processes, such as cancer pathogenesis. Identifying these partners is essential for understanding the intricate molecular mechanisms that govern cellular functions. Tools and servers such as STRING,[14] KEGG,[15] and REACTOME[16] provide valuable resources to unravel these interactions and uncover the network of relationships within a pathway. STRING is a powerful bioinformatics resource that specializes in predicting protein–protein interactions. It integrates various sources of experimental and computational data to construct a comprehensive network of functional associations between proteins. KEGG is a widely used resource for understanding biological pathways and the interactions among genes and proteins in various organisms. REACTOME is another valuable resource for pathway analysis, providing a curated knowledge base of biological pathways. Consolidating all the results and identifying the potential functional partner will be subjected to further computational analysis.
Protein–Protein Docking
The protein–protein docking between two or more proteins was performed using the HADDOCK 2.4 server, an advanced computational tool specifically designed for modeling macromolecular interactions.[17] The main objective of this docking study was to predict the potential binding modes and interface interactions between these two important proteins, which play crucial roles in signaling pathways and cellular processes. Parameters were set to define ERBB2 as the “receptor” and the identified functional protein as the “ligand,” given their respective roles in the interaction.
Active residues, crucial for the protein–protein interaction, were defined based on the known literature and biological context. These active residues were set to be unbound and flexible during the docking simulations. The initial docking runs were performed using the rigid body docking mode, allowing for a global search of possible binding orientations. HADDOCK generated an ensemble of docking solutions for further refinement. The top-scoring docking solutions were selected for the semiflexible refinement stage. During this step, the side chains of the active residues were allowed to optimize their positions, employing a simulated annealing protocol. The resulting docked complexes were analyzed to identify the most probable binding mode, interface residues, hydrogen bonding, and hydrophobic interactions between ERBB2 and functional proteins.[5] [18] The binding energy of the top-ranked solution was used as an indicator of the stability of the predicted complex.
Toxicity Prediction Analysis
The toxicity prediction of the identified lead compounds was performed using the ProTox-II server, a widely recognized computational tool specifically designed for predicting the potential toxicity of small molecules.[19] This step was crucial in the drug discovery process to assess the safety profile of the identified lead compounds before further experimental investigations. The top five potential lead compounds were selected based on their favorable binding energies and predicted binding modes from our structure-based virtual screening analysis. ProTox-II provided predictions for multiple toxicity endpoints, including mutagenicity, hepatotoxicity, carcinotoxicity, and others, using validated models. The predicted toxicity scores were interpreted, considering both the individual toxicity endpoints and the overall toxicity profile.[20] The toxicity predictions were critically analyzed in the context of the intended therapeutic application and the known safety standards for pharmaceutical compounds. This approach ensures that the lead compounds with the most promising efficacy and favorable safety profiles are advanced in the drug discovery pipeline, enhancing the overall success rate of the drug development process.
Ethics
No human participants/subjects were involved in this study.
Results
Virtual Screening Studies
The structure-based drug design study conducted using AutoDock Vina yielded promising results in the search for potential drug candidates from the Enamine and LifeChemicals databases. After rigorous filtering based on the Lipinski Rule of Five, which ensures drug-likeness and favorable pharmacokinetic properties, a total of 385,000 and 137,000 compounds were retained from the LifeChemicals and Enamine databases, respectively, for further analysis. The AutoDock Vina program automatically generates the grid map and presents clustered results to users in a transparent manner. Within Vina, diverse stochastic global optimization techniques, including genetic algorithms, simulated annealing, and particle swarm optimization, were employed. The active site cavity was carefully chosen, followed by postdocking steps involving energy minimization and H-bond optimization.
Upon thorough docking simulations, we identified 10 compounds that exhibited notably favorable binding energies, suggesting strong interactions with the receptor ([Table 1]). The binding energy calculated for the top compounds ranged from –8.346 to –6.296 kcal/mol. The interacting residues were Thr5, Arg412, Leu414, and Ser441 with the docked ligands ([Fig. 1]). These compounds demonstrated specific amino acid interactions within the binding site or active site of the receptor, reinforcing the potential for selective binding and biological activity. The identification of these 10 compounds with both promising binding energy and significant interactions with the receptor represents a significant outcome of our study.
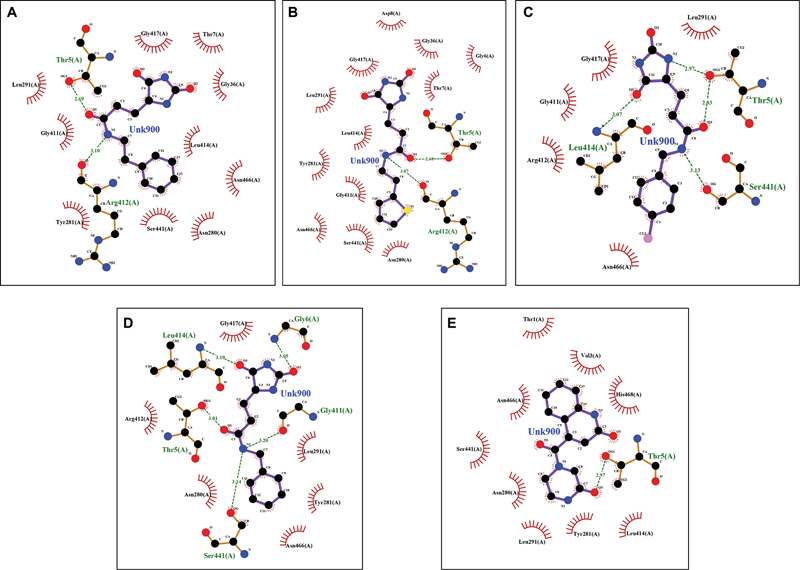
| | Fig 1Two-dimensional interaction diagram of the top five compounds in the active site of the protein. (A) LC_87763; (B) LC_33378; (C) LC_27122; (D) Enamine_101102; and (E) LC_87632.|
From the results we observe, most of the potential compounds were identified from the LifeChemicals database. Out of the 10 compounds, 7 from LifeChemicals and 3 are from the Enamine database. The identified compounds were found to have hydrogen bond, salt-bridge, and pi–pi interactions.
Protein Functional Partners
The comprehensive analysis of functional partners using STRING, KEGG, and REACTOME has yielded crucial insights into the intricate network of interactions involving the ERBB2 protein. Through STRING, we uncovered a multitude of potential interaction partners, which were further enriched and contextualized within biological pathways and processes using the KEGG and REACTOME databases. The rigorous exploration highlighted a significant finding: among the various candidates, epidermal growth factor receptor (EGFR) emerged as the most prominent and compelling functional partner for the ERBB2 protein ([Fig. 2]). This outcome was supported by multiple lines of evidence, including high-confidence protein–protein interaction scores, shared pathways, and known biological relevance. The identification of EGFR as the best functional partner of ERBB2 underscores its central role in cellular signaling and its potential significance in various physiological and pathological contexts. Further experimental validation and functional studies will be crucial to decipher the specific mechanisms through which this interaction contributes to cellular processes and disease pathways, potentially opening new avenues for therapeutic interventions targeting the ERBB2–EGFR complex.
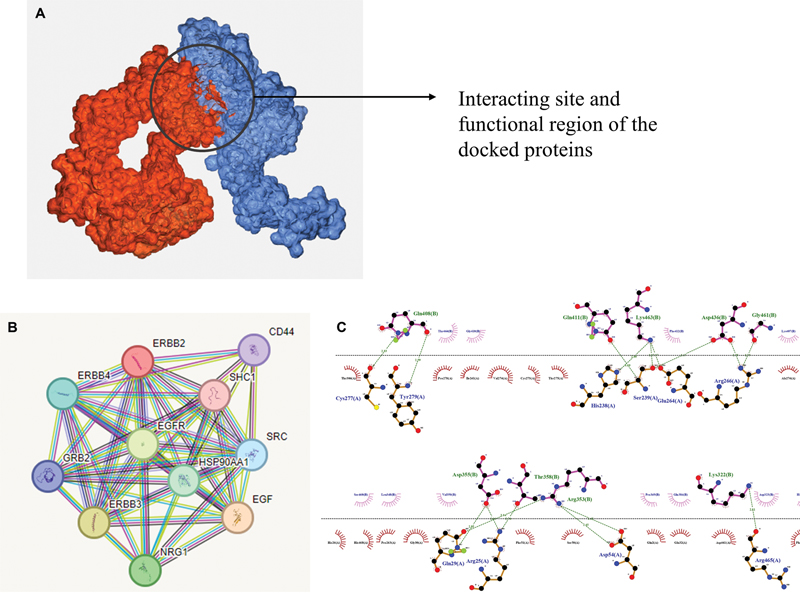
| Fig 2 (A) Protein–protein docking interaction and molecule binding pose representing the interaction site. (B) Interacting amino acid residues from chain A (ERBB2) and chain B (epidermal growth factor receptor).|
Protein–Protein Docking
The protein–protein docking studies conducted using HADDOCK 4.2 server provided critical insights into the binding interactions between ERBB2 as the receptor protein and EGFR as the ligand. The docking simulations yielded a range of potential binding modes, allowing us to explore the diverse conformations in which these two proteins may interact. Through comprehensive analysis, we identified a highly favorable binding mode that demonstrated strong binding energy, indicative of stable and specific interactions between ERBB2 and EGFR ([Table 2]). The best cluster is observed as Cluster 1 with a Haddock score of –95.3, lowest root mean square deviation (RMSD) of 0.6 Å, –384 kcal/mol of Electrostatic energy, and –95.9 kcal/mol of Van der Walls energy. The structure selected from the initial cluster exhibits a Haddock score of –91.46, a minimal RMSD of 0.4 Å, and recorded energy values of –311 kcal/mol for Electrostatic forces and –90.2 kcal/mol for Van der Waals interactions. The important residues of EGFR found in/around the active site comprises nearly 30 amino acids from 353 to 359 and 448 to 464; specifically, Gln408, Lys463, Phe412, and Asp436 are vital residues. The detailed examination of the docked complex revealed key amino acid residues involved in the binding interface, highlighting the precise molecular interactions contributing to the formation of the ERBB2–EGFR complex ([Fig. 2]). These findings shed light on the potential functional implications of this protein–protein interaction, further underscoring the significance of the ERBB2–EGFR interaction in cellular signaling pathways and disease contexts.
|
Parameters |
Values |
|---|---|
|
Cluster size |
53 |
|
Haddock score |
–95.3 ± 12.5 |
|
Van der Waals energy |
–95.9 ± 5.1 |
|
Electrostatic energy |
–384.2 ± 42.6 |
|
RMSD from the overall lowest-energy structure |
0.6 ± 0.4 |
|
Desolvation energy |
0.0 ± 3.3 |
|
Restraints violation energy |
774.2 ± 27.8 |
|
Buried surface area |
3,259.9 ± 151.7 |
|
Z-score |
–2.1 |
References
- Ashraf SQ, Nicholls AM, Wilding JL, Ntouroupi TG, Mortensen NJ, Bodmer WF. Direct and immune mediated antibody targeting of ERBB receptors in a colorectal cancer cell-line panel. Proc Natl Acad Sci U S A 2012; 109 (51) 21046-21051
- Pectasides E, Bass AJ. ERBB2 emerges as a new target for colorectal cancer. Cancer Discov 2015; 5 (08) 799-801
- Gilbertson RJ. ERBB2 in pediatric cancer: innocent until proven guilty. Oncologist 2005; 10 (07) 508-517
- Hynes NE. ErbB2: from an EGFR relative to a central target for cancer therapy. Cancer Res 2016; 76 (13) 3659-3662
- Sivaiah G, Raveesha R, Prasad SBB. et al. Synthesis, anticancer activity and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives as EGFR/HER2 dual kinase inhibitors. J Mol Struct 2023; 1289
- Bateman A, Martin MJ, Orchard S. et al; UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2021; 49 (D1): D480-D489
- Loganathan L, Natarajan K, Muthusamy K. Computational study on cross-talking cancer signalling mechanism of ring finger protein 146, AXIN and Tankyrase protein complex. J Biomol Struct Dyn 2020; 38 (17) 5173-5185
- Loganathan L, Briget Kuriakose B, Lobelle Sampayan E, Muthusamy K. Targeting renin receptor for the inhibition of renin angiotensin aldosterone system: an alternative approach through in silico drug discovery. Comput Theor Chem 2022; 1208: 113541
- Loganathan L, Gopinath K, Sankaranarayanan VM. et al. Computational and pharmacogenomic insights on hypertension treatment: rational drug design and optimization strategies. Curr Drug Targets 2020; 21 (01) 18-33
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 2010; 24 (05) 417-422
- Li Q, Shah S. Structure-based virtual screening. Methods Mol Biol 2017; 1558: 111-124
- Shaikh N, Linthoi RK, Swamy KV, Karthikeyan M, Vyas R. Comprehensive molecular docking and dynamic simulations for drug repurposing of clinical drugs against multiple cancer kinase targets. J Biomol Struct Dyn 2023; 41 (16) 7735-7743
- Loganathan L, Muthusamy K. Investigation of drug interaction potentials and binding modes on direct renin inhibitors. A computational modeling studies. Lett Drug Des Discov 2018; 15
- Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 2000; 28 (18) 3442-3444
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999; 27 (01) 29-34
- Croft D, Mundo AF, Haw R. et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014; 42 (Database issue, D1): D472-D477
- van Zundert GCP, Rodrigues JPGLM, Trellet M. et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol 2016; 428 (04) 720-725
- Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release 2010; 146 (03) 264-275
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2018; 46 (W1): W257-W263
-
Raies AB,
Bajic VB.
In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci 2016; 6 (02) 147-172
Address for correspondence
Abdulhadi Almazroea, MDAssociate Professor of General Pediatrics, Medical College, Taibah UniversityMedina 42353Saudi ArabiaEmail: Abdalhadi66@yahoo.comEmail: amazroea@taibahu.edu.saPublication History
Article published online:
02 September 2024© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, IndiaWe recommend- Discovery of Tyrosinase Inhibitors: Structure-Based Virtual Screening and Biological EvaluationPharmaceutical Fronts, 2022
- Search for cannabinoid receptor 1 antagonists using structure-based virtual screening: identification of natural product hitsP Pandey, Planta Medica, 2014
- Search for cannabinoid receptor 1 antagonists using structure-based virtual screening: identification of natural product hitsP Pandey, Planta Medica, 2014
- Neurologic Manifestations of Pediatric Acquired Immunodeficiency Syndrome: Clinical Features and Therapeutic ApproachesMark Mintz, Seminars in Neurology, 1992
- Neurologic Manifestations of Pediatric Acquired Immunodeficiency Syndrome: Clinical Features and Therapeutic ApproachesMark Mintz, Seminars in Neurology, 1992
- Traditional and Machine Learning Approaches in Structure-Based Drug Virtual ScreeningHong Zhang, Chinese Journal of Chemical Physics, 2024
- Target Molecular-Based Neuroactivity Screening and Analysis of Panax ginseng by Affinity Ultrafiltration, UPLC-QTOF-MS and Molecular DockingLide Yu, The American Journal of Chinese Medicine, 2019
- Dendritic Cell-Based Cancer Immunotherapy Targeting Wilms’ Tumor 1 for Pediatric CancerShigetaka Shimodaira, Exon Publications, 2016
- Introduction to Mentalization-Based Approaches for Parents, Children, Youths, and FamiliesJana Volkert, American Journal of Psychotherapy, 2021
- Porphyrinoid-based photosensitizers for diagnostic and therapeutic applications: An updateAmit Aggarwal, Journal of Porphyrins and Phthalocyanines, 2019
- Discovery of Tyrosinase Inhibitors: Structure-Based Virtual Screening and Biological Evaluation
| | Fig 1Two-dimensional interaction diagram of the top five compounds in the active site of the protein. (A) LC_87763; (B) LC_33378; (C) LC_27122; (D) Enamine_101102; and (E) LC_87632.|
| Fig 2 (A) Protein–protein docking interaction and molecule binding pose representing the interaction site. (B) Interacting amino acid residues from chain A (ERBB2) and chain B (epidermal growth factor receptor).|
References
- Ashraf SQ, Nicholls AM, Wilding JL, Ntouroupi TG, Mortensen NJ, Bodmer WF. Direct and immune mediated antibody targeting of ERBB receptors in a colorectal cancer cell-line panel. Proc Natl Acad Sci U S A 2012; 109 (51) 21046-21051
- Pectasides E, Bass AJ. ERBB2 emerges as a new target for colorectal cancer. Cancer Discov 2015; 5 (08) 799-801
- Gilbertson RJ. ERBB2 in pediatric cancer: innocent until proven guilty. Oncologist 2005; 10 (07) 508-517
- Hynes NE. ErbB2: from an EGFR relative to a central target for cancer therapy. Cancer Res 2016; 76 (13) 3659-3662
- Sivaiah G, Raveesha R, Prasad SBB. et al. Synthesis, anticancer activity and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives as EGFR/HER2 dual kinase inhibitors. J Mol Struct 2023; 1289
- Bateman A, Martin MJ, Orchard S. et al; UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2021; 49 (D1): D480-D489
- Loganathan L, Natarajan K, Muthusamy K. Computational study on cross-talking cancer signalling mechanism of ring finger protein 146, AXIN and Tankyrase protein complex. J Biomol Struct Dyn 2020; 38 (17) 5173-5185
- Loganathan L, Briget Kuriakose B, Lobelle Sampayan E, Muthusamy K. Targeting renin receptor for the inhibition of renin angiotensin aldosterone system: an alternative approach through in silico drug discovery. Comput Theor Chem 2022; 1208: 113541
- Loganathan L, Gopinath K, Sankaranarayanan VM. et al. Computational and pharmacogenomic insights on hypertension treatment: rational drug design and optimization strategies. Curr Drug Targets 2020; 21 (01) 18-33
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 2010; 24 (05) 417-422
- Li Q, Shah S. Structure-based virtual screening. Methods Mol Biol 2017; 1558: 111-124
- Shaikh N, Linthoi RK, Swamy KV, Karthikeyan M, Vyas R. Comprehensive molecular docking and dynamic simulations for drug repurposing of clinical drugs against multiple cancer kinase targets. J Biomol Struct Dyn 2023; 41 (16) 7735-7743
- Loganathan L, Muthusamy K. Investigation of drug interaction potentials and binding modes on direct renin inhibitors. A computational modeling studies. Lett Drug Des Discov 2018; 15
- Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 2000; 28 (18) 3442-3444
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999; 27 (01) 29-34
- Croft D, Mundo AF, Haw R. et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014; 42 (Database issue, D1): D472-D477
- van Zundert GCP, Rodrigues JPGLM, Trellet M. et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol 2016; 428 (04) 720-725
- Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release 2010; 146 (03) 264-275
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2018; 46 (W1): W257-W263
- Raies AB, Bajic VB. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci 2016; 6 (02) 147-172