 PDF
PDF  Views
Views  Share
Share


Spectrum of Somatic Malignancy in Testicular Germ Cell Tumors—A Histopathological Review of 25 Cases with Clinical Outcome
CC BY 4.0 · Indian J Med Paediatr Oncol 2025; 46(02): 174-182
DOI: DOI: 10.1055/s-0044-1788307
Abstract
Introduction : Germ cell tumors (GCTs) are the commonest testicular malignancy in young males. These tumors are highly chemoresponsive, however become resistant to conventional therapy when a somatic-type malignancy (SM) develops, which happens in ∼3 to 6% of the cases.
Materials and Methods : We reviewed the histologic profile of all cases of testicular/retroperitoneal GCT with SM, diagnosed over a period of 12 years in our institute. Correlation of histologic profile with clinical outcome was done wherever feasible.
Results : A total of 25 cases of testicular/retroperitoneal GCT with SM were identified for review. The histological spectrum of SMs included carcinoma (n = 9), sarcoma (n = 9), embryonic-type neuroectodermal tumor (ENET) (n = 4), and other rare histological types (n = 3). SMs were frequently seen at the resected metastatic sites (n = 13) and in postchemotherapy setting (n = 12); 14 cases had concurrent GCT and SM at the time of diagnosis/initial resection and 9 cases presented as late relapses (more than 2 years after initial presentation). Four patients were treated with metastasectomy and lymph node dissection, six patients were treated with combined resection and chemotherapy, and nine patients were treated with only adjuvant chemotherapy. The patients with SM confined to testis and those treated with multimodality approach had relatively better outcome.
Conclusion : GCTs with SM are a highly heterogeneous group of tumors with varying histologic types and management strategies. Strict adherence to histological diagnostic criteria, differentiating these tumors from close mimics such as glandular and sarcomatoid yolk sac tumors, teratomatous overgrowth, and a new second primary somatic tumor are important due to implications in management and prognosis.
Keywords
Publication History
14 August 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
- Embryogenesis and Metastatic Testicular Germ Cell Tumors of AdolescentsL. H. J. Looijenga, Klinische Pädiatrie, 2009
- Germ cell tumors of the ovary : a review of 48 casesU.DBAFNA R PALLAVI, Indian J Radiol Imaging, 2004
- Tumor Marker Decline in Predicting Treatment Outcome among Poor-Risk Testicular Germ Cell Tumors—A Tertiary Cancer Center dataLakshmi Haridas K., South Asian Journal of Cancer
- Mediastinal Germ Cell TumorsAlan Sandler, Seminars in Respiratory and Critical Care Medicine, 1997
- Intracranial Germ Cell TumorsTeri Danielle You Ying Yeoh, Seminars in Neurology
- Primary Mediastinal Germ Cell Tumors in Children and Adolescents: a report of 6 cases<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Hypersensitivity pneumonitis: Clinical spectrum and outcome<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Modeling Clinical Outcome of Children With Autistic Spectrum Disorders<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Clinical spectrum and outcome of non IPF patients with a typical UIP picture<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Long-term Outcome and Clinical Spectrum of 73 Pediatric Patients With Mitochondrial Diseases<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
Abstract
Introduction : Germ cell tumors (GCTs) are the commonest testicular malignancy in young males. These tumors are highly chemoresponsive, however become resistant to conventional therapy when a somatic-type malignancy (SM) develops, which happens in ∼3 to 6%. of the cases.
Materials and Methods : We reviewed the histologic profile of all cases of testicular/retroperitoneal GCT with SM, diagnosed over a period of 12 years in our institute. Correlation of histologic profile with clinical outcome was done wherever feasible.
Results : A total of 25 cases of testicular/retroperitoneal GCT with SM were identified for review. The histological spectrum of SMs included carcinoma (n = 9), sarcoma (n = 9), embryonic-type neuroectodermal tumor (ENET) (n = 4), and other rare histological types (n = 3). SMs were frequently seen at the resected metastatic sites (n = 13) and in postchemotherapy setting (n = 12); 14 cases had concurrent GCT and SM at the time of diagnosis/initial resection and 9 cases presented as late relapses (more than 2 years after initial presentation). Four patients were treated with metastasectomy and lymph node dissection, six patients were treated with combined resection and chemotherapy, and nine patients were treated with only adjuvant chemotherapy. The patients with SM confined to testis and those treated with multimodality approach had relatively better outcome.
Conclusion : GCTs with SM are a highly heterogeneous group of tumors with varying histologic types and management strategies. Strict adherence to histological diagnostic criteria, differentiating these tumors from close mimics such as glandular and sarcomatoid yolk sac tumors, teratomatous overgrowth, and a new second primary somatic tumor are important due to implications in management and prognosis.
Keywords
Introduction
Testicular tumors are rare, accounting for 1 to 2%. of all tumors in males.[1] Germ cell tumors (GCTs) are the commonest testicular tumors (98%) in young males.[2] Despite being highly malignant tumors, most are amenable to chemotherapy (70–80% cure rate after first-line chemotherapy)[3] and hence, curable (95% 5-year survival rate).[4] [5] [6] About 3 to 6%. of GCTs develop somatic-type malignancy (SM),[7] which is defined as the occurrence of a distinct component of somatic-type malignant neoplasm, as seen in other organs. The SM component may be epithelial or mesenchymal, and is diagnosed when it measures 5 mm or more with an infiltrative or expansile growth pattern.[8] [9] Various histological types of SM have been described—sarcoma (commonly rhabdomyosarcoma [RMS]), adenocarcinoma, peripheral neuroectodermal tumor (PNET) (presently renamed embryonic-type neuroectodermal tumor [ENET] in World Health Organization [WHO] 2022), nephroblastoma, neuroblastoma, and other rare tumor types.[10]
SM occurs frequently in relapses and following chemotherapy but may be present at the time of initial presentation infrequently.[10] It is an important cause of resistance to conventional platinum-based chemotherapeutic regimens.[11] It confers poor prognosis when seen in metastatic sites.[4]
Though studies on therapeutic strategies and clinical outcome of this disease are available, literature on detailed histopathological features is limited to a few case series and multi-institutional studies. Herein, we provide a detailed review of pathological features of 25 cases of GCT with SM diagnosed at our institute.
Materials and Methods
Primary Outcome
To study the clinicopathological features of testicular GCT with SM.
Secondary Outcome
To correlate the histological factors with clinical characteristics of the patients.
Case Selection
The surgical pathology database was retrospectively searched for all cases of primary testicular/retroperitoneal GCTs with SM diagnosed over a period of 12 years (January 2011–June 2023), including in-house and referral cases. The histopathology slides and relevant immunohistochemical stains performed at the time of initial diagnosis were reviewed.
Inclusion Criteria
Only cases confirming to the WHO diagnostic criteria for SM were included in the study (malignant nodules with infiltrative or expansile growth measuring at least 5 mm).
Exclusion Criteria
Cases with teratomatous overgrowth and cases with scattered atypical stromal cells/small atypical epithelial cell clusters were excluded.
Parameters Studied
For cases with unequivocal diagnosis of SM, histological type of SM was assigned as per WHO 2022 terminology. Other histological features such as presence of associated GCT components were noted. The demographic details, clinical presentation, treatment, and outcome details were collected from the institutional electronic medical records database. Histological type of SM, site, and timescale of occurrence (SM occurring concurrently with GCT at initial presentation or presentation as late relapse) were compared with clinical parameters and outcome in patients with adequate available clinical information.
Statistical Analysis
The data collected were entered in MS Excel and the mean, percentage, and statistical analysis were performed by SPSS version 20.1.
Ethical Statement
Institutional Ethics Committee II, Institutional Review Board, Tata Memorial Center, Mumbai. All procedures performed in this study involving human particiapants were in accordance with the ethical standards of the Institutional Ethics Committee II, Institutional Review Board, Tata Memorial Center, Mumbai and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Results
About 1,650 cases of testicular GCTs were diagnosed in the study period at our institute, of which 25 cases had developed SM (1.52%). A total of 28 cases were initially diagnosed as GCTs with SM; however, on review of histopathological features, only 25 cases met the defined criteria for SM and 3 cases were excluded (teratomatous [glandular and rhabdomyomatous] overgrowth [n = 2] and seminoma with anaplastic features [n = 1]).
Clinical Parameters
The median age at diagnosis of SM was 31.96 years with an age range of 17 to 48 years. All patients had primary testicular GCT (24/25) except one case of primary retroperitoneal GCT (1/25). SM was detected only in primary site in 11 cases (44%) (testis n = 10, retroperitoneum n = 1), only in metastatic sites in 12 cases (48%), and in both testis and retroperitoneal lymph nodes (RPLNs) in 2 cases. The metastatic sites were RPLN (10/25), left supraclavicular lymph nodes (1/25), and lung (1/25). The median age of patients with SM in primary site only was 27 and that of patients with SM in metastatic sites was 32.5 years.
About 14 cases (56%) had concurrent GCT and SM at the time of initial presentation and 9 cases (36%) presented as late relapses (at least 2 years after initial presentation). About half of the patients (n = 12, 48%) had received prior chemotherapy and/or radiation therapy. The mean time to development of SM in metastatic sites from the time of primary GCT diagnosis was 3.5 years (range 0–22 years).
Pathological Parameters
At initial presentation, 21 cases had nonseminomatous GCT (NSGCT), 2 cases had mixed GCT, 1 case had a burnt-out GCT with germ cell neoplasia in situ (GCNIS); 1 case showed only SM (ENET) in the limited sections available (sections from adjacent testis was not available for review). Concomitant GCT was noted along with SM in 19 cases; teratomatous component was present in 18/19 cases; 1 case had only yolk sac tumor (YST) (1/19); 1 case had only GCNIS. In five cases, no residual GCT component was identified at the time of diagnosis of SM, of which three cases had only core biopsy material for evaluation.
In primary site (testis/retroperitoneum), the histotypes encountered in decreasing order of frequency are sarcoma (6/11; 54.55%), ENET (2/11; 18.18%), mucinous adenocarcinoma (2/11; 18.18%), and Wilms' tumor (1/11; 9.09%). In metastatic sites, the histological types encountered in decreasing order of frequency are carcinoma (7/12; 58.33%), sarcoma (3/12; 25%), ENET (1/12; 8.33%), and desmoplastic small round cell tumor (DSRCT) (1/12; 8.33%). The two cases with presence of SM concurrently in testis and RPLN were carcinosarcoma—with adenocarcinoma and spindle cell sarcoma not otherwise specified (NOS) areas and a case of ENET with scattered SALL4-positive large cells (discussed in detail in the following section).
The histological types of SM observed were carcinoma (9/25; 36%), sarcoma (9/25; 36%), ENET (4/25; 16%), 1 case each of carcinosarcoma (1/25; 4%), Wilms' tumor (1/25; 4%), and DSRCT (1/25; 4%). Sarcoma (RMS) was the most common type of SM in testis and carcinoma (adenocarcinoma) was the most common pathology in metastatic sites. SM which presented as late relapses were adenocarcinoma (7/9), ENET (1/4), sarcoma NOS (1/9), and RMS (1/9).
Among carcinomas, adenocarcinoma was the commonest histotype (7/9); one case had mucinous histology and remaining were adenocarcinoma, NOS ([Fig. 1]). Two cases in this group had unusual histology—neuroendocrine carcinoma (NEC) and clear cell renal cell carcinoma (CCRCC). The tumor was present only in a small focus in RPLN and measured only 0.5 to 1 cm in both these cases. In the case of NEC, SM was detected 1 year after initial diagnosis of GCT; the tumor showed nested and pseudoglandular patterns with marked nuclear atypia and brisk mitotic activity. It was immunopositive for synaptophysin, CD56, and chromogranin while being negative for EMA, glypican 3, CD30, and cKIT. The patient was given chemotherapy (VIP regimen) and is disease free, 4 years after diagnosis. The case of CCRCC was detected 3 years after the initial diagnosis of GCT; the SM showed classical morphology of CCRCC as seen in kidney with low nuclear grade ([Fig. 1]). The tumor cells were positive for AE1/AE3, PAX8, and CD10; negative for TFE3 and HMB45. The patient was thoroughly investigated for any occult primary in kidney and no renal mass was found on radiological evaluation. No adjuvant second-line chemotherapy was given and the patient is disease free, 5 years after diagnosis. Both these cases had residual teratomatous elements in the RPLND along with SM.
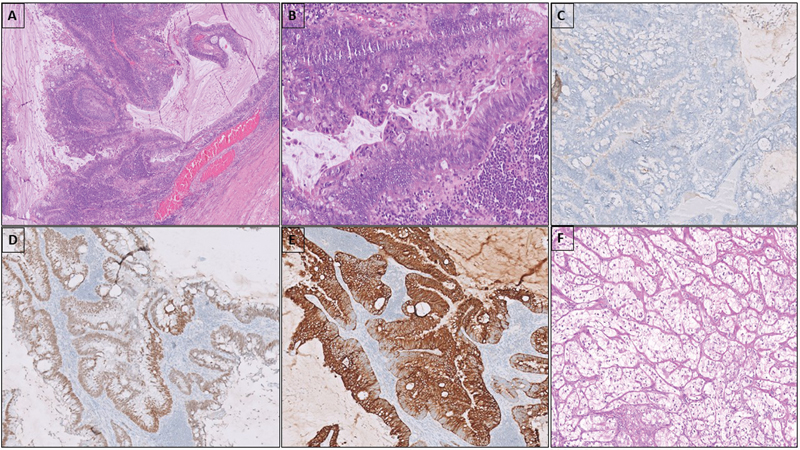
Fig 1: Adenocarcinoma with mucinous columnar cells in glandular and papillary architecture (A). The cells show nuclear stratification and atypia with frequent mitoses (B). Tumor cells are negative for SALL4 (C), positive for SATB2 (D) and CK20 (E). The case of clear cell renal cell carcinoma composed of nests of clear cells with low nuclear grade (F).
Among the sarcoma group, seven cases were RMS (7/9) and two cases were high-grade spindle cell sarcoma, NOS (2/9). All RMSs were of embryonal subtype with an appreciable number of rhabdomyoblasts; showed positivity for desmin (focal), myogenin, and/or myoD1.
The two sarcoma, NOS cases did not show any line of differentiation by immunohistochemistry (smooth muscle, skeletal muscle, and neural lineage markers were negative). The cases showed sheets and vague fascicles of spindle cells with significant nuclear atypia and frequent mitoses ([Fig. 2]). The tumor was small in one case, measuring 0.8 cm in diameter and was confined to testis; the patient underwent RPLND and no adjuvant chemotherapy was given and patient is disease free 6 months after diagnosis. The other case presented as late relapse in RPLN had disease progression despite second-line chemotherapy.
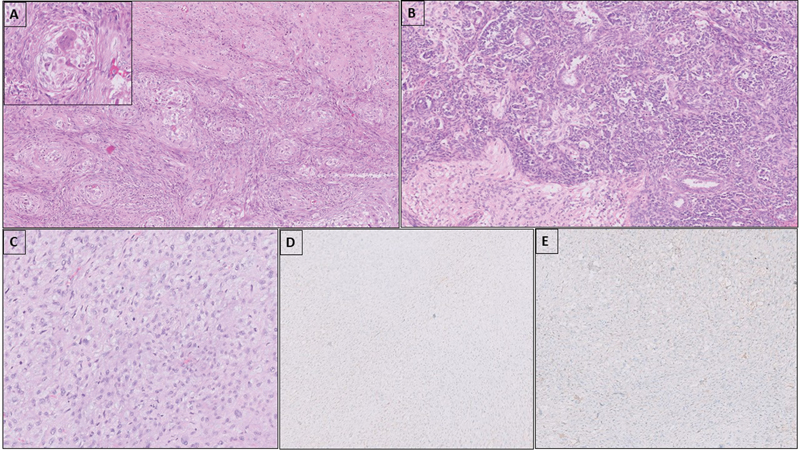
Fig 2 : Carcinosarcoma with nests of clear epithelioid cells and atypical spindle cells around the nests (A). Central blood vessel and multinucleate giant cell can be seen in the center of the epithelioid cell nests (inset). Teratoid Wilms' tumor with nests and sheets of blastemal cells, epithelial tubular elements, and intimately admixed mature glial tissue (B). Sarcoma, NOS with sheets of mitotically active atypical spindle cells (C); the tumor cells are negative for glypican 3 (D) and desmin (E).
In the ENET group (n = 4), the tumor was confined to testis in two cases; was seen in both testis and RPLN in one case and in the other case, it was seen as a late relapse in RPLN. The tumor cells were positive for SOX11 and negative for SALL4 and YST markers; showed variable immunopositivity for synaptophysin, chromogranin, CD56, and S100. NKX2–2 and GFAP were negative in the cases tested ([Fig. 3]). EWSR1 break-apart fluorescence in situ hybridization (FISH) was negative in one case tested for. All except one patient responded well to chemotherapy.
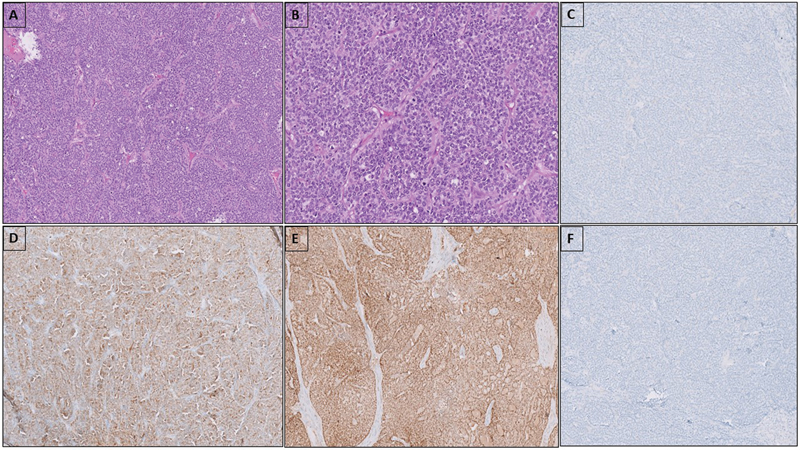
Fig 3 : Embryonic-type neuroectodermal tumor composed of sheets and nests of monomorphic round cells with scant cytoplasm and fine granular chromatin (A, B). Tumor cells are negative for NKX2–2 (C), diffusely positive for S100 (D) and SOX11 (E). Staining for OCT 3/4 is negative (F).
Cases with Unusual Histological Features
The case of carcinosarcoma showed intimately admixed epithelial and sarcomatous components, seen in primary site and RPLN. In testis, sarcomatous component was dominant, composed of sheets and vague fascicles of atypical spindle cells with frequent mitosis along with small nests of epithelial cells with clear cytoplasm. At places epithelial cells were seen condensed around thin-walled blood vessels ([Fig. 2]). In RPLN, epithelial component was prominent and showed features of adenocarcinoma, NOS with infiltrating, irregular angulated atypical glands. Immunostains for SALL4, OCT3/4, desmin, CD34, and inhibin were negative in both epithelial and spindle cell components. The residual GCT component present was a small focus of YST in the RPLN.
In the case of triphasic Wilms' tumor we encountered, there was intimately admixed glial elements, seen along with NSGCT elements, the SM being present at primary site at the time of initial presentation. The tumor showed a predominant blastemal component along with intermingled islands of teratoid/glial elements (mature and immature) in a neuropil-rich matrix ([Fig. 2]).
The case of DSRCT was detected in RPLNs, 2 years following the occurrence of GCT. The tumor showed malignant round cell morphology with fibrous stroma and a polyphenotypic immunoprofile. The tumor cells were immunopositive for AE1/AE3, desmin, WT1, synaptophysin, MIC-2, and FLI1. Reverse transcriptase polymerase chain reaction for EWSR1-WT1 and EWSR1-FLI1 gene fusions, both were negative.
An intriguing histological finding we observed in a case of ENET was presence of singly scattered large cells with prominent nuclei amidst malignant round blue cells. The smaller cells were positive for synaptophysin, SALL4, and NKX2.2, and negative for desmin, myogenin, calretinin, and D2–40; the scattered large cells were positive for SALL4, D2–40, and OCT3/4. The tumor had mature teratomatous elements and GCNIS was noted in the adjacent testicular parenchyma. There was no seminoma/YST components. This unusual occurrence of admixed large cells in ENET has not been observed previously, to the best of our knowledge. Considering their immunophenotype we could best regard these cells to be entrapped GCNIS cells.
Outcome Details
Clinical follow-up details were available for 19 patients, 18 patients were alive and 1 was deceased, with mean follow-up duration being 4.88 years (range 0.5–17 years).
Treatment details following the diagnosis of SM were available for 20 patients; 9 patients received only chemotherapy; 5 patients underwent surgical and medical (chemotherapy) management; 4 patients underwent surgical management only; 1 patient was treated with combined chemotherapy and radiotherapy; 1 patient received chemotherapy, radiotherapy, and surgery.
One patient died of surgical complications (post-RPLND) and all other patients were alive for the available follow-up period. Nine patients were disease free and asymptomatic; four patients did not respond well to second-line chemotherapy and had been put on palliative intent management, one patient had stable residual disease after treatment and was being followed up, and four patients had not completed treatment at the time of last follow-up.
Among the patients who had disease progression after second-line therapy (4/19), two were sarcoma (2/4), one DSRCT (1/4), and one ENET (1/4). Other than the testicular ENET, three cases had SM at metastatic sites.
Among the patients treated with surgical resection alone (n = 4) or combined surgical resection and chemotherapy/radiotherapy (n = 7), most showed good response to treatment (8/11) and were disease free; two patients (DSRCT and RMS; both detected in metastatic sites) developed progressive disease despite multimodality management and one patient (RMS detected in RPLN) died due to surgery-related complications. Among the patients treated only with chemotherapy (n = 9), two patients developed progressive disease (sarcoma NOS and ENET with large cells) and one patient had residual disease (adenocarcinoma) after the completion of chemotherapy; three patients are yet to complete the chemotherapy course; two patients were disease free after the completion of treatment; outcome details are not available for a patient.
Discussion
Occurrence of SMs in GCT was described as early as 1946 and included in WHO 2004 under the term “teratoma with somatic-type malignancy”; later renamed “GCTs with somatic type malignancy.”[12] SM is believed to arise from pluripotent germ cells or malignant transformation of teratomatous/yolk-sac elements. Some studies have reported occurrence of SM after chemotherapy, which may be due to its DNA damaging effect.[10] [13] Pathogenetically, SMs retain the characteristic molecular abnormality, isochromosome 12p seen in postpubertal-type GCTs which can be detected as 12p amplification by FISH.[7]
SMs are most frequently encountered in metastatic sites as primary treatment failure or late disease relapses.[14] [15] This finding was reiterated in our study as in nearly 56%. of cases SM was seen in metastatic sites and more than half of the patients had received prior chemotherapy. The overall prevalence of SM was less in our cohort than the 3 to 6%. prevalence observed in most other studies. More cases were identified in the past 5 years in our cohort (17/25), probably due to better understanding of this entity and clearer diagnostic criteria. The histological spectrum of SM described in GCT is diverse (sarcomas, carcinomas of various phenotypes, PNET, glial tumors, and nephroblastomas), sarcomas being the commonest in most series (63%).[5] In our series, we observed an equal occurrence of carcinoma and sarcoma with the next frequent being ENET; some rare tumor types such as nephroblastoma, CCRCC, carcinosarcoma, DSRCT, and neuroendocrine carcinoma were also encountered.
We observed carcinomas more frequently in metastatic sites as late relapses, the commonest type being adenocarcinoma, similar to the observation in various studies.[16] The CCRCC and neuroendocrine carcinoma cases we encountered have been not or only sparsely described in literature, respectively. There has been a single report of papillary renal cell carcinoma arising as SM in RPLNs and no other additional renal tumor types have been described previously.[12] [17] Both these tumors were present only in a small focus in our cases, measuring <1>
The commonest type of sarcoma in our series was embryonal RMS, as reported in studies previously. During the initial screening, we found two cases of rhabdomyomatous overgrowth misdiagnosed as RMS. Rhabdomyomatous overgrowth has been described following chemotherapy and is composed of sheets of maturing skeletal muscle cells without any malignant round cell areas, necrosis, or apoptosis. Hence, detecting immature rhabdomyoblasts or embryonal areas is needed to ascertain a diagnosis of RMS as a SM. In our study, among the RMS group, all except one case showed good response to chemotherapy, with disease-free interval of 1 to 57 months.
The sarcoma, NOS cases did not show any line of differentiation by immunohistochemistry. The closest differential to be considered in such cases is sarcomatoid YST. The sarcomatoid areas in YST can be low or high grade,[18] usually show foci with epithelioid pattern and are immunopositive for SALL4 and glypican 3. Sarcomatoid YST is resistant to platinum-based chemotherapy and has an aggressive clinical course, much like GCT with SM.[14]
The so-called testicular PNETs are distinct from the PNET/Ewings sarcoma of bone and soft tissue. The testicular PNETs are negative for FLI1 and CD99; do not show EWSR1 gene rearrangements, a hallmark of PNET. These tumors are generally confined to testis, frequently contain areas resembling medulloepithelioma, medulloblastoma, and a range of neuroglial tumors.[14] [19] Therefore, these tumors are considered to represent “central” rather than peripheral-type neuroectodermal tumors and have been renamed ENET in the WHO fifth edition.[8] [20] ENETs when confined to testis are increasingly being recognized to have better outcome. In comparison, metastatic ENET may have a poorer outcome, as seen in one of our cases which progressed despite second-line chemotherapy.
One of our cases was diagnosed as DSRCT based on histological and immunohistochemical features, although molecular findings were not supportive. Occurrence of this tumor as a SM has not been described previously.
Most of the SMs are consistently associated with teratoma, hence the need to carefully distinguish these tumors from immature teratomatous elements, benign teratomatous overgrowth, and teratomas showing nuclear atypia. Teratomatous overgrowth and atypia in stromal cells are well known to occur after chemotherapy. Any form of teratomatous overgrowth can mimic disease progression, presenting with increasing size of the mass clinically; however, it does not behave in a malignant fashion. In our review, two cases initially misdiagnosed as SM were reclassified as teratomatous overgrowth after histological review.
As somatic malignant transformation is common in metastatic sites and can present as late relapses,[16] metastasis from a new primary nongerm cell neoplasm becomes a diagnostic consideration. In such instances, young age of the patient, prior history of GCT, presence of 12p gain, and absence of primary tumor after systematic evaluation points to a diagnosis of SM.[5]
The various histotypes of SM and relevant clinical parameters described in the literature with significant case numbers are summarized in [Table 1]. In most studies, poor clinical outcome of GCT with SM was observed only when SM involved metastatic sites.[14] In our series too, most of the patients with SM confined to testis had better outcome with only one patient found to have residual disease posttreatment. Also, some studies have questioned the strict size-based diagnostic criteria for SM, as a case with small focus of ENET (not meeting the 5 mm criteria) presented later with widespread disease.[7] In our series, three cases which had SM occupying less than 1 cm area in slide showed a good outcome, despite the aggressive histological type (neuroendocrine carcinoma, CCRCC, and high-grade sarcoma).
|
Our study |
Magers et al (2014)[5] |
Lobo et al (2022)[7] |
Hwang et al (2022)[4] |
Scheckel et al (2019)[21] |
Sharma et al (2019)[10] |
|
|---|---|---|---|---|---|---|
|
Study overview |
Morphology, immunophenotype and outcome |
Morphology and immunophenotype |
Histological spectrum and molecular features |
Histological type and clinical outcome |
Management and clinical outcome |
Management and clinical outcome |
|
Study period |
12 y |
23 y, single institution |
16 y, two institutions |
15 y, single institution |
35 y, single institution |
18 y, single institution |
|
Number of cases |
25 |
124 cases of testicular GCT (excluded PNET and Wilms) 84 histologically confirmed as SM after IHCs |
30 (testicular and primary retroperitoneal GCT) |
63 (excluded sarcomatoid YST) |
24 (testicular, mediastinal and pineal) |
30 |
|
No. of cases with SM in testis |
13 |
4 |
14 |
22 |
2 |
– |
|
No. of cases with SM in metastatic sites |
14 |
– |
22 |
41 |
22 |
– |
|
Presence of associated GCT |
All except two (one showed only GCNIS and other showed only ENET) |
Initial diagnosis of GCT known in 50% patients |
Initial diagnosis of GCT established in all cases |
All except one case of ENET |
– |
– |
|
Concomitant initial presentation of SM with GCT |
14 |
7 cases |
14 (5 were ENET) |
– |
12 |
9 |
|
H/O chemotherapy and/or radiation therapy before SM |
9 |
83% |
15 |
35 |
21 |
|
|
Carcinoma |
9 (adenocarcinoma, clear cell RCC, neuroendocrine carcinoma) |
44 (adenocarcinoma, carcinoma NOS and sarcomatoid carcinoma) |
11 (including papillary RCC, neuroendocrine carcinoma) |
21 (adenocarcinoma, carcinoma NOS, neuroendocrine carcinoma) |
12 (adenocarcinomas) |
3 |
|
Sarcoma |
9 (RMS, sarcoma NOS) |
39 cases (RMS, myxofibrosarcoma, sarcoma NOS, UPS, LMS, OGS, gliosarcoma) |
6 |
21 (RMS, angiosarcoma, sarcoma NOS) |
12 (RMS and sarcoma, NOS) |
16 |
|
ENET |
4 (one with scattered SALL4-positive large cells) |
– |
8 |
15 |
– |
10 |
|
Other rare tumor types |
Two cases of carcinosarcoma, one case of DSRCT |
High-grade glioma |
Neuroblastoma, low-grade glial tumor |
Nephroblastoma, mixed RMS, and small cell carcinoma |
– |
Nephroblastoma |
|
IHC |
Relevant markers pertaining to histotype |
Reclassification of 37 cases as glandular/sarcomatoid YST |
– |
NA |
– |
– |
|
Commonest histological type in metastatic sites |
Carcinoma |
– |
Adenocarcinoma |
Carcinoma |
– |
– |
|
Molecular studies |
– |
NA |
12p FISH, EWSR1 rearrangement in ENET |
NA |
– |
– |
|
Clinical outcome |
Carcinoma frequent in late relapses, ENET and sarcoma frequent at initial presentation; SM at metastasis—poor outcome; small SM size (<1> |
Sarcomatoid SM more aggressive than glandular SM |
Late relapse, high-grade sarcomatous SM, ENET, extragonadal primary predictive of poor outcome |
Poor survival in metastatic SM group and carcinoma histological type |
Carcinomas had better overall survival compared with sarcoma group |
Late relapse had poor outcome |
References
- Park JS, Kim J, Elghiaty A, Ham WS. Recent global trends in testicular cancer incidence and mortality. Medicine (Baltimore) 2018; 97 (37) e12390
- Ghazarian AA, Trabert B, Devesa SS, McGlynn KA. Recent trends in the incidence of testicular germ cell tumors in the United States. Andrology 2015; 3 (01) 13-18
- Országhová Z, Kalavska K, Mego M, Chovanec M. Overcoming chemotherapy resistance in germ cell tumors. Biomedicines 2022; 10 (05) 972
- Hwang MJ, Hamza A, Zhang M. et al. Somatic-type malignancies in testicular germ cell tumors: a clinicopathologic study of 63 cases. Am J Surg Pathol 2022; 46 (01) 11-17
- Magers MJ, Kao CS, Cole CD. et al. “Somatic-type” malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am J Surg Pathol 2014; 38 (10) 1396-1409
- Gillessen S, Sauvé N, Collette L. et al; International Germ Cell Cancer Classification Update Consortium. Predicting outcomes in men with metastatic nonseminomatous germ cell tumors (NSGCT): results from the IGCCCG Update Consortium. J Clin Oncol 2021; 39 (14) 1563-1574
- Lobo J, Rodrigues Â, Henrique R. et al. Morphological spectrum and molecular features of somatic malignant transformation in germ cell tumours. Histopathology 2022; 81 (01) 84-98
- Idrees MT, Looijenga LH, Boormans JL, Colecchia M. Teratoma with somatic-type malignancy. In: Urinary and Male Genital Tumours. WHO Classification of Tumours Editorial Board. Lyon (France): International Agency for Research on Cancer. 5th ed; 2022
- Ibrahim DY, Sun H. Somatic malignant transformation of a testicular teratoma: a case report and an unusual presentation. Case Rep Pathol 2019; 2019: 5273607
- Sharma A, Alifrangis C, Milic M. et al. Somatic transformation in metastatic testicular germ cell tumours - a different disease entity. Anticancer Res 2019; 39 (09) 4911-4916
- Malagón HD, Valdez AM, Moran CA, Suster S. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 2007; 31 (09) 1356-1362
- Mikuz G, Colecchia M. Teratoma with somatic-type malignant components of the testis. A review and an update. Virchows Arch 2012; 461 (01) 27-32
- Bahrami A, Ro JY, Ayala AG. An overview of testicular germ cell tumors. Arch Pathol Lab Med 2007; 131 (08) 1267-1280
- Guo CC, Czerniak B. Somatic-type malignancies in testicular germ cell tumors. Hum Pathol 2022; 127: 123-135
- Washino S, Konishi T, Saito K, Ohshima M, Nakamura Y, Miyagawa T. Two cases of somatic-type malignancy as a very late relapse of testicular cancer successfully managed by surgical resection. J Surg Case Rep 2017; 2017 (11) rjx233
- Kranendonk MEG, Hackeng WM, Offerhaus GJA. et al. The decisive role of molecular pathology in presumed somatic metastases of type II testicular germ cell tumors: report of 2 cases. Diagn Pathol 2020; 15 (01) 99
- Zeh N, Wild PJ, Bode PK. et al. Retroperitoneal teratoma with somatic malignant transformation: a papillary renal cell carcinoma in a testicular germ cell tumour metastasis following platinum-based chemotherapy. BMC Urol 2013; 13: 9
- Acosta AM, Al-Obaidy KI, Sholl LM. et al. Sarcomatoid yolk sac tumor harbors somatic mutations that are otherwise rare in testicular germ cell tumors. Am J Surg Pathol 2022; 46 (05) 701-712
- Murati Amador B, Matoso A. Testicular germ cell tumor showing concurrent PNET and neuroglial neoplasms with wide spectrum of grades. Am J Surg Pathol 2019; 43 (06) 865-867
- Bhoopathi HK, Tanveer N, Naskar S, Gautum HV. Testicular PNET arising in a mixed germ cell tumor: a diagnosis not to be missed. Indian J Surg Oncol 2021; 12 (03) 637-640
- Scheckel CJ, Kosiorek HE, Butterfield R, Ho TH, Hilal T. Germ cell tumors with malignant somatic transformation: a Mayo Clinic experience. [published correction appears in Oncol Res Treat 2019;42(6):354] Oncol Res Treat 2019; 42 (03) 95-100
Address for correspondence
Publication History
Article published online:
14 August 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
- Embryogenesis and Metastatic Testicular Germ Cell Tumors of AdolescentsL. H. J. Looijenga, Klinische Pädiatrie, 2009
- Germ cell tumors of the ovary : a review of 48 casesU.DBAFNA R PALLAVI, Indian J Radiol Imaging, 2004
- Tumor Marker Decline in Predicting Treatment Outcome among Poor-Risk Testicular Germ Cell Tumors—A Tertiary Cancer Center dataLakshmi Haridas K., South Asian Journal of Cancer
- Mediastinal Germ Cell TumorsAlan Sandler, Seminars in Respiratory and Critical Care Medicine, 1997
- Mediastinal Germ Cell TumorsAlan Sandler, Semin Respir Crit Care Med, 1997
- Testicular torsion: clinical analysis of 16 cases<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Torsion of testicular appendage: clinical analysis of 16 cases<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Rosacea and Its Association With Malignancy: Systematic Review<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Clinical analysis of 7 cases with IgG 4-related disease misdiagnosed as malignant tumors<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
- Identification of Intracranial Germ Cell Tumors Based on Facial Photo: Software Development Using Deep Learning Technology<svg viewBox="0 0 24 24" fill="none" xmlns="http://www.w3.org/2000/svg">
Fig 1: Adenocarcinoma with mucinous columnar cells in glandular and papillary architecture (A). The cells show nuclear stratification and atypia with frequent mitoses (B). Tumor cells are negative for SALL4 (C), positive for SATB2 (D) and CK20 (E). The case of clear cell renal cell carcinoma composed of nests of clear cells with low nuclear grade (F).
Fig 2 : Carcinosarcoma with nests of clear epithelioid cells and atypical spindle cells around the nests (A). Central blood vessel and multinucleate giant cell can be seen in the center of the epithelioid cell nests (inset). Teratoid Wilms' tumor with nests and sheets of blastemal cells, epithelial tubular elements, and intimately admixed mature glial tissue (B). Sarcoma, NOS with sheets of mitotically active atypical spindle cells (C); the tumor cells are negative for glypican 3 (D) and desmin (E).
Fig 3 : Embryonic-type neuroectodermal tumor composed of sheets and nests of monomorphic round cells with scant cytoplasm and fine granular chromatin (A, B). Tumor cells are negative for NKX2–2 (C), diffusely positive for S100 (D) and SOX11 (E). Staining for OCT 3/4 is negative (F).
References
- Park JS, Kim J, Elghiaty A, Ham WS. Recent global trends in testicular cancer incidence and mortality. Medicine (Baltimore) 2018; 97 (37) e12390
- Ghazarian AA, Trabert B, Devesa SS, McGlynn KA. Recent trends in the incidence of testicular germ cell tumors in the United States. Andrology 2015; 3 (01) 13-18
- Országhová Z, Kalavska K, Mego M, Chovanec M. Overcoming chemotherapy resistance in germ cell tumors. Biomedicines 2022; 10 (05) 972
- Hwang MJ, Hamza A, Zhang M. et al. Somatic-type malignancies in testicular germ cell tumors: a clinicopathologic study of 63 cases. Am J Surg Pathol 2022; 46 (01) 11-17
- Magers MJ, Kao CS, Cole CD. et al. “Somatic-type” malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am J Surg Pathol 2014; 38 (10) 1396-1409
- Gillessen S, Sauvé N, Collette L. et al; International Germ Cell Cancer Classification Update Consortium. Predicting outcomes in men with metastatic nonseminomatous germ cell tumors (NSGCT): results from the IGCCCG Update Consortium. J Clin Oncol 2021; 39 (14) 1563-1574
- Lobo J, Rodrigues Â, Henrique R. et al. Morphological spectrum and molecular features of somatic malignant transformation in germ cell tumours. Histopathology 2022; 81 (01) 84-98
- Idrees MT, Looijenga LH, Boormans JL, Colecchia M. Teratoma with somatic-type malignancy. In: Urinary and Male Genital Tumours. WHO Classification of Tumours Editorial Board. Lyon (France): International Agency for Research on Cancer. 5th ed; 2022
- Ibrahim DY, Sun H. Somatic malignant transformation of a testicular teratoma: a case report and an unusual presentation. Case Rep Pathol 2019; 2019: 5273607
- Sharma A, Alifrangis C, Milic M. et al. Somatic transformation in metastatic testicular germ cell tumours - a different disease entity. Anticancer Res 2019; 39 (09) 4911-4916
- Malagón HD, Valdez AM, Moran CA, Suster S. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 2007; 31 (09) 1356-1362
- Mikuz G, Colecchia M. Teratoma with somatic-type malignant components of the testis. A review and an update. Virchows Arch 2012; 461 (01) 27-32
- Bahrami A, Ro JY, Ayala AG. An overview of testicular germ cell tumors. Arch Pathol Lab Med 2007; 131 (08) 1267-1280
- Guo CC, Czerniak B. Somatic-type malignancies in testicular germ cell tumors. Hum Pathol 2022; 127: 123-135
- Washino S, Konishi T, Saito K, Ohshima M, Nakamura Y, Miyagawa T. Two cases of somatic-type malignancy as a very late relapse of testicular cancer successfully managed by surgical resection. J Surg Case Rep 2017; 2017 (11) rjx233
- Kranendonk MEG, Hackeng WM, Offerhaus GJA. et al. The decisive role of molecular pathology in presumed somatic metastases of type II testicular germ cell tumors: report of 2 cases. Diagn Pathol 2020; 15 (01) 99
- Zeh N, Wild PJ, Bode PK. et al. Retroperitoneal teratoma with somatic malignant transformation: a papillary renal cell carcinoma in a testicular germ cell tumour metastasis following platinum-based chemotherapy. BMC Urol 2013; 13: 9
- Acosta AM, Al-Obaidy KI, Sholl LM. et al. Sarcomatoid yolk sac tumor harbors somatic mutations that are otherwise rare in testicular germ cell tumors. Am J Surg Pathol 2022; 46 (05) 701-712
- Murati Amador B, Matoso A. Testicular germ cell tumor showing concurrent PNET and neuroglial neoplasms with wide spectrum of grades. Am J Surg Pathol 2019; 43 (06) 865-867
- Bhoopathi HK, Tanveer N, Naskar S, Gautum HV. Testicular PNET arising in a mixed germ cell tumor: a diagnosis not to be missed. Indian J Surg Oncol 2021; 12 (03) 637-640
- Scheckel CJ, Kosiorek HE, Butterfield R, Ho TH, Hilal T. Germ cell tumors with malignant somatic transformation: a Mayo Clinic experience. [published correction appears in Oncol Res Treat 2019;42(6):354] Oncol Res Treat 2019; 42 (03) 95-100