 PDF
PDF  Views
Views  Share
Share


Should We Look beyond Revised International Prognostic Scoring System: A Retrospective Observational Study of Progression of Myelodysplastic Syndrome to Acute Leukemia
CC BY-NC-ND 4.0 ? Indian J Med Paediatr Oncol 2021; 42(05): 431-438
DOI: DOI: 10.1055/s-0041-1736175
Abstract
Introduction?Myelodysplastic syndrome (MDS) is a clonal stem cell disorder and heterogeneous condition resulting in peripheral cytopenias with marrow dysplasia due to ineffective hematopoiesis. The revised International Prognostic Scoring System (IPSS-R) predicts the risk of progression to acute leukemia (AL). Indian data on MDS and its progression to AL are limited. Additionally, the cytogenetic findings are dictated by patients' racial background. Study intended to analyze the cytogenetic profile of the patients with MDS.
Objectives?This study aimed to (1) evaluate the clinicohematologic and morphologic spectrum of newly diagnosed MDS cases, (2) evaluate the cytogenetic profile of these cases, and (3) study the cases progressed to AL.
Materials and Methods?MDS cases diagnosed and followed-up during a 5-year study period, from January 2015 to December 2019, were included in the study and the study was conducted at regional cancer center in Western India. De novo diagnosed MDS cases with complete workup were considered and MDS due to secondary causes were excluded. Baseline clinical, hematologic findings were tabulated along with cytogenetics and risk stratified as per IPSS-R, and their progression was studied.
Results?A total of 63 cases of de novo MDS were diagnosed over a period of 5 years with 45 cases on follow-up and 15 cases (33.3%) progressed to AL. Maximum number of cases belonged to MDS-excess blast (EB) category accounting to 48 cases (76.1%). Apparently normal karyotyping was the commonest cytogenetic finding in 33 MDS cases (61.2%) and in 8 cases that progressed to AL (53.4%).
Conclusion?MDS cases diagnosed at relatively early age were at higher risk of progression to AL. Majority of the cases that progressed to AL were risk stratified in high and very high risk groups and 10 cases which progressed to AL belonged to good category, interestingly apparent normal karyotyping was the commonest cytogenetic finding in more than 50% of the cases progressed to AL. Molecular mutations could only explain this progression and studies integrating molecular mutations with present IPSS-R scoring system should be conducted, as it could translate into better risk stratification and help in early identification and better management of cases at risk in progression to AL.
Keywords
myelodysplastic syndrome - acute leukemia - International prognostic scoring system - cytogeneticsEthics
The procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1964, as revised in 2013. The Gujarat Cancer & Research Institute (GCRI) ethical committee board approval was obtained, no: IRC/2021/P-14 on March 19, 2021.
Since the study was retrospective, informed consent was not required and the study did not include any intervention. Waiver of informed consent was obtained from the Ethics Committee.
Abstract
Introduction?Myelodysplastic syndrome (MDS) is a clonal stem cell disorder and heterogeneous condition resulting in peripheral cytopenias with marrow dysplasia due to ineffective hematopoiesis. The revised International Prognostic Scoring System (IPSS-R) predicts the risk of progression to acute leukemia (AL). Indian data on MDS and its progression to AL are limited. Additionally, the cytogenetic findings are dictated by patients' racial background. Study intended to analyze the cytogenetic profile of the patients with MDS.
Objectives?This study aimed to (1) evaluate the clinicohematologic and morphologic spectrum of newly diagnosed MDS cases, (2) evaluate the cytogenetic profile of these cases, and (3) study the cases progressed to AL.
Materials and Methods?MDS cases diagnosed and followed-up during a 5-year study period, from January 2015 to December 2019, were included in the study and the study was conducted at regional cancer center in Western India. De novo diagnosed MDS cases with complete workup were considered and MDS due to secondary causes were excluded. Baseline clinical, hematologic findings were tabulated along with cytogenetics and risk stratified as per IPSS-R, and their progression was studied.
Results?A total of 63 cases of de novo MDS were diagnosed over a period of 5 years with 45 cases on follow-up and 15 cases (33.3%) progressed to AL. Maximum number of cases belonged to MDS-excess blast (EB) category accounting to 48 cases (76.1%). Apparently normal karyotyping was the commonest cytogenetic finding in 33 MDS cases (61.2%) and in 8 cases that progressed to AL (53.4%).
Conclusion?MDS cases diagnosed at relatively early age were at higher risk of progression to AL. Majority of the cases that progressed to AL were risk stratified in high and very high risk groups and 10 cases which progressed to AL belonged to good category, interestingly apparent normal karyotyping was the commonest cytogenetic finding in more than 50% of the cases progressed to AL. Molecular mutations could only explain this progression and studies integrating molecular mutations with present IPSS-R scoring system should be conducted, as it could translate into better risk stratification and help in early identification and better management of cases at risk in progression to AL.
Keywords
myelodysplastic syndrome - acute leukemia - International prognostic scoring system - cytogeneticsIntroduction
Myelodysplastic syndrome (MDS) is a clonal hematological disorder with ineffective hematopoiesis in one or more bone marrow (BM) lineages.[1] Clinically MDS presents with fatigue, dyspnea, infections, easy bruising due to dysplasia in cell lines, and peripheral cytopenias. BM failure occurs due to ineffective hematopoiesis as a result of excessive apoptosis, maturation arrest, and proliferation resembling the mechanism that play in acute myeloid leukemia (AML).[2] Multiple factors are implicated in pathophysiology of MDS such as immunologic, cytogenetic, epigenetic, genetic factors, and therapy associated factors.[3]
MDS progress to secondary AML (sAML) in 20 to 30% of cases, whereas the remaining succumb to progressive BM failure.[4] The prognosis of patients transforming to AML is generally grave, as they are resistant to currently available treatment options and the long-term survival rate among treated patients is low.[5]
Owing to the inherent nature of genetic heterogeneity, multiple cytogenetic abnormalities are detected; however, role of each of these in disease progression is not well established.[6] Risk assessment and therapeutic planning is presently based on the revised International Prognostic Scoring System (IPSS-R).[7] Cytogenetics (CG), one of the parameters in IPSS-R, is performed mostly by conventional CG. Based on the scoring system, the risk of transformation to AML is assessed and managed accordingly. With advent of sequencing, genetic information obtained can greatly impact the prognosis of the disease.[8]
Deep sequencing studies has facilitated in better understanding of molecular pathogenesis of MDS and its progression. Majority of the mutations do not seem to play any causative role other than genetic instability.[6] [9] Disease progression of MDS is characterized by increasing blast count which is due to acquired additional mutations and genetic alterations in the new emerging clones.[9] This explains the varied mutational profiles across the different stages of disease.
The model to understand MDS and its progression into AML is a multistep concept of leukemogenesis[10] [11] which is characterized by accumulation of different molecular and genomic alterations belonging to class-I and -II genes and epigenetic modifications[12] [13] that result in the expansion of the MDS/AML clone, this is well understood and accepted.
Spectrum of some mutated genes enriched in sAML is?FLT3,?NRAS,?WT1,?IDH1, and?IDH2?(type-I genes) which are also the most frequently mutated genes in primary AML.[14] The commonly mutated genes in high-risk MDS group are?GATA2,?RUNX1,?TET2,?TP53, and?ASXL1?(type-II genes).[15] Leukemic transformation is faster in patients with type-I gene mutations than those without type-I mutations. Epigenetic and RNA splicing genes are commonly mutated in MDS and acquisition of type-I mutations, providing proliferation advantage and explaining the multistep concept of leukemogenesis.[16]
Around 80 to 90% of MDS cases have somatic mutations identified which determine the clinical phenotype and overall survival. Sequencing studies help to identify mutations which are predictors of poor overall survival.[10] These mutations are not commonly assessed during patient management as definitive guidelines are not well established. Currently the Indian data on MDS cases and its progression to AL is limited. The study was performed to evaluate clinicohematologic, morphological spectrum, and cytogenetic findings of newly diagnosed MDS along with outcome and also evaluate the cases which progressed to AL with emphasis on their cytogenetic findings.
Materials and Methods
Observational study was done in a retrospective manner and was conducted in patients diagnosed with MDS at a regional cancer center in western India, from January 2015 to December 2019. Sample size included all the cases diagnosed at our institute over a period of 5 years. Cases which were not evaluated for CG and secondary causes of MDS were excluded. Cases were diagnosed if they met the established criteria, diagnosis of de novo MDS was considered following correlation with morphologic, cytogenetic, and clinical criteria. Cytopenia was diagnosed as per the recommended and established thresholds in IPSS-R (hemoglobin concentration <10 xss=removed>9/L); BM examination which is considered hallmark in diagnosis of MDS was evaluated for morphologic dysplasia in all the three cell lineages and the established cut-off was 10% in each lineage to call unilineage or multilineage dysplasia. Dysplastic changes observed in nucleus of erythroid lineage (nuclear budding, internuclear bridging, karyorrhexis, multinuclearity, and megaloblastoid changes) and in cytoplasm (ringed sideroblasts with an aid of Perl's stain and cytoplasmic vacoulations). Dysgranulopoiesis is characterized by small or overtly large cell size, nuclear hypo- or hypersegmentation, decreased granularity, and auer rods. Dysplastic changes in megakaryocytes include micromegakaryocytes, nuclear hypolobation, and multinucleation. Immunohistochemistry was performed to correlate with BM biopsy findings in necessary cases. Biochemical tests such as vitamin B-12, folate, and iron profile were performed in all cases to rule out nutritional causes of dysplasia. Any occupational exposure to heavy metals or pesticides and anticancer drugs was excluded. Cases without workup for secondary causes of dysplasia and prior history of treatment for any other malignancy were excluded.
Conventional cytogenetics: BM aspirate sample was sent in heparinized tube for CG. Karyotyping was performed using short-term culture technique and metaphase chromosomes were banded by Giemsa- trypsin- guanine cytosine (GTG) technique and documented as per International System for Human Cytogenetic Nomenclature (ISCN) 2016 guidelines.[17] At least 20 normal metaphases were necessary to consider a patient cytogenetically normal.
Fluorescence in situ hybridization (FISH): interphase and metaphase FISH was performed and tests were performed as per clinician request. Slides were prepared by phase-contrast microcopy and probes were added to target area of interest. Slides were subjected to denaturation and hybridization process, counterstained with 4',6-diamidino-2-phenylindole (DAPI), and visualized under fluorescence microscope.
FISH was performed with commercially available probes (Vysis), the following probes were used: LSI 5q31 (EGR1) and 7q31 (D7S486). Probe of interest was simultaneously applied (cohybridized) with a different color-labeled probe (internal control). The combinations used were 5q31 (spectrum orange) and D5S721:D5S23 (spectrum green), 7q31 (spectrum orange), and CEP 7 (spectrum green). Cells were considered harboring a gene deletion when carrying two signals due to the internal control and only one signal due to the target sequence and monosomic when carrying one signal each for the internal control and target sequence probes. A total of 200 cells were analyzed and cut-off was 2% for del 7q and 4% for del 5q.
Diagnosis was based on the World Health Organization (WHO) 2016 criteria and cases prior to this were reevaluated and classified as per the present criteria. With the necessary data, the patients were risk stratified as per IPSS-R system.[7] During the follow-up period, the cases which progressed to AL were identified using the WHO determined blast cut-off of 20%.
Statistical Analysis
Statistical analysis was performed with Statistical Package for Social Science (SPSS) software version 20 (IBM SPSS Statistics for Windows, Armonk, New York, United States; IBM Corp.). Continuous variables were reported as median with standard deviation (SD) and categorical variables as percentages. The association between categorical variables was evaluated for significance, period of progression to AL was calculated in the each risk group and Student's?t-test was applied to study the significance of age and its progression also among different risk categories of MDS. The test was considered significant if the?p-value is <0>
Results
Over a period of 5 years, 63 cases of primary MDS were reported (45 men and 18 women) and out of 45 cases on follow-up, 15 cases (33.3%) progressed to AL during the median follow-up period of 6 months (range: 1?53 months). Median age at diagnosis of primary MDS is 52 years (range: 11?79 years) and median age of cases which progressed to AL is 51 years. Among the cases that progressed to AL, 53 years (range: 11?74 years) was the median age in men (11 cases) and 56 years (range: 51?71 years) in women (4 cases). Most patients complained of fatigue (58 cases [92.0%]), breathlessness on exertion (33 cases [52.4%]), bleeding or easy bruising was presenting complaint (7 seven cases [11.1%]), six cases (9.5%) were incidentally detected due to cytopenia/s which was unresponsive to therapy, and two asymptomatic cases (3.1%) were also referred due to presence of atypical cells/blasts in peripheral blood.
Of the 63 patients, 38 cases (71.9%) had hemoglobin concentration of <8>10?g/dL. Mean hemoglobin concentration (MHC) was 7.4?g/dL (SD???0 1.74). Absolute neutrophil count (ANC) less than 800/?L was seen in 21 cases (31.7%) and remaining case had count more than 800/?L. Overall, 31 cases (49.2%) had platelet count <50 href="https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0041-1736175#SM21490073-1" xss=removed>Supplementary Material S1], available in the online version), and 12 cases (19.0%) had isolated anemia and 29 cases (46.0%) were pancytopenic. Macrocytic morphology was the commonest finding followed by dimorphic picture and in three cases, microcytic red blood cell (RBC) was noted. Eight cases had blasts in peripheral blood. Vitamin B-12 and serum folate levels evaluated were within normal limits and iron profile in few cases revealed an overload picture.
The distribution of WHO defined MDS entities along with median age in each of the entity is enumerated in ([Table 1]). Maximum number of cases belonged to MDS-excess blast (EB) EB2 category accounted to 32 cases (50.7%). Cytogenetic study results are available in 54 cases and it includes all cases which progressed to AL. Twenty-one cases (38.9%) had abnormal karyotype and the CG findings in decreasing order of their prevalence in the study is normal karyotyping (33 cases), del (7q; 8 cases), del (5q; 5 cases), monosomy 7 (3 cases; [Fig. 1]) trisomy 8 (3 cases), complex karyotyping +der (7) del(7) i(17; 1 case), and hyperdiploidy (1 case; [Table 2]). Risk groups stratified by IPSS-R and median age in each of the risk groups is tabulated ([Table 3]). Forty-five cases were on follow-up and 15 cases (33.3%) progressed to AL which included one case of acute lymphoblastic leukemia (ALL; [Table 4]). Estimated EFS was 5 months (95% confidence interval [CI]: 0?12 months), 17 cases were on lenalidomide and 5 cases (29.4%) progressed to AML over 6 months, and 7 out of 13 cases (53.4%) on hypomethylating agents (HMA) transformed to AML over 6 months. Six cases were on blood transfusion and four cases were on erythropoietin, one case each progressed to AML over 11 and 12 months, respectively. Patient aged 35 years with 15% blast and marrow fibrosis was treated as AML, 11 months of follow-up period was uneventful. Hematopoietic stem cell transplantation (HSCT) was performed in an 18-year-old patient belonging high-risk category with normal karyotyping, patient progressed to AML in 48 months and is on follow-up from past 12 months. Patient aged 11 years who progressed to ALL after 4 months of diagnosis of MDS with del 7q and is on follow-up from past 53 months and is stable. All cases which progressed to AL belonged to MDS-EB category only and majority of them fell into high and very-high-risk IPSS-R groups ([Table 2]). Median time taken to progress to AL in high and very high risk groups was 11 and 6 months, respectively, while longer time duration of 16 months was taken by intermediate risk group. Progression to AL in high (p?=?0.01) and very high risk group (p?=?0.002) was statistically significant, while intermediate risk group was insignificant (p?=?0.3). Monosomy 7 belonging to poor cytogenetic risk group, showed 100% transformation to AL. One case with complex karyotyping was lost for follow-up.
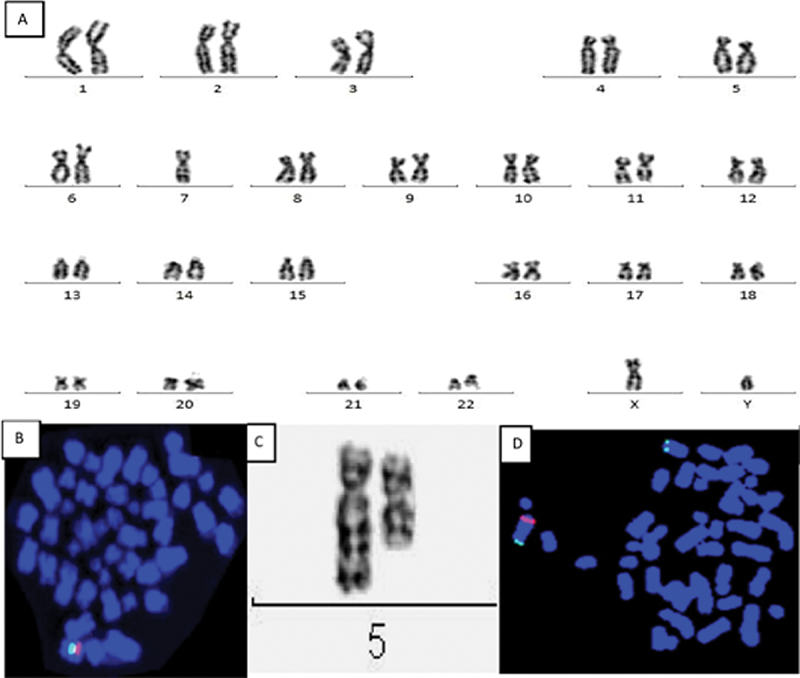
|?Fig. 1(A)?Complete karyogram shows 45,XY,-7 indicating complete loss of chromosome 7. (B) Metaphase FISH shows orange signal localized to short arm of chromosome 7 (7q31) region and control probe targeting the centromere CEP7 shows green signal. Only one green and orange signal (101G) seen indicates Monosomy 7. (C) Partial karyotype shows interstitial deletion in long arm of chromosome 5. In del(5q), breakpoint occur in q14 and q33. (D) FISH probe localized to short arm of chromosome 5 (5q31) generates orange signal and control probe localized to long arm (5p15.2) and generates green signal. Metaphase FISH shows one orange signal and two green signal (102G) indicating deletion 5q.|
|
Age |
Sex |
HB (g/dL) |
ANC |
Platelet |
PS (blast) |
Blast % (marrow) |
Dysplastic cells |
MDS subtype |
Cytogenetics |
Risk group |
Treatment |
Time to progress |
Progressed to |
Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
(y) |
(/?L) |
(/?L) |
(mo) |
(mo) |
||||||||||
|
42 |
M |
8.6 |
0.6 |
0.21 |
0 |
11 |
EGM |
EB2 |
NK |
VH |
HMA |
13 |
AML |
LFU |
|
62 |
F |
8.6 |
2.6 |
0.95 |
0 |
9 |
EGM |
EB1 |
NK |
H |
HMA |
19 |
AML |
Death |
|
69 |
F |
7.3 |
2.3 |
0.91 |
0 |
16 |
G |
EB2 |
del 5q |
VH |
HMA |
5 |
AML |
4 |
|
56 |
M |
10.1 |
3.5 |
0.05 |
0 |
6 |
EGM |
EB1 |
NK |
I |
Lena[a] |
20 |
AML |
5 |
|
11 |
M |
4.3 |
1.1 |
0.34 |
8 |
16 |
GM |
EB2 |
del 7q |
VH |
HMA[b] |
3 |
ALL |
53 (on follow up) |
|
36 |
M |
8.6 |
4.1 |
0.19 |
0 |
5 with auer rods |
EM |
EB2 |
NK |
H |
Lena |
6 |
AML |
LFU |
|
51 |
F |
6.9 |
1.2 |
1.3 |
0 |
7 |
E |
EB1 |
NK |
H |
EPO[c] |
11 |
AML |
3 |
|
71 |
F |
9.1 |
0.7 |
0.51 |
0 |
6 |
EGM |
EB1 |
NK |
H |
Lena |
28 |
AML |
5 |
|
65 |
M |
7.7 |
1.5 |
0.35 |
5 |
15 |
EGM |
EB2 |
Monosomy7 |
VH |
HMA |
1 |
AML |
Death |
|
71 |
M |
8.7 |
20.4 |
1.54 |
11 |
13 |
E |
EB2 |
Monosomy7 |
VH |
HMA |
3 |
AML |
3 |
|
74 |
M |
10 |
10.1 |
2.98 |
0 |
5 |
E |
EB1 |
NK |
I |
BT |
12 |
AML |
11 |
|
31 |
M |
8.4 |
0.7 |
0.31 |
0 |
11 |
EGM |
EB2 |
Monosomy7 |
VH |
Lena |
6 |
AML |
13 |
|
53 |
M |
7 |
0.1 |
0.26 |
0 |
12 |
EGM |
EB2 |
del 5q |
VH |
Lena |
1 |
AML |
3 |
|
18 |
M |
6.6 |
10 |
0.45 |
0 |
5 |
EG |
EB2 |
NK |
H |
HSCT |
48 |
AML |
12 (on follow-up) |
|
40 |
M |
7.6 |
0.2 |
0.91 |
0 |
14 |
G |
EB2 |
trisomy 8 |
VH |
HMA |
1 |
AML |
LFU |
- Tefferi A, Vardiman JW.?Myelodysplastic syndromes. N Engl J Med 2009; 361 (19) 1872-1885
- Devita Jr. VT, Lawrence TA, Rosenberg SA.?DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology. 9th ed.. Philadelphia, PA: Wolters Kluwer; 2011
- Visconte V, Selleri C, Maciejewski JP, Tiu RV.?Molecular pathogenesis of myelodysplastic syndromes. Transl Med UniSa 2014; 8 (04) 19-30
- Steensma DP, Bennett JM.?The myelodysplastic syndromes: diagnosis and treatment. Mayo Clin Proc 2006; 81 (01) 104-130
- Haase D.?Cytogenetic features in myelodysplastic syndromes. Ann Hematol 2008; 87 (07) 515-526
- Shukron O, Vainstein V, K?ndgen A, Germing U, Agur Z.?Analyzing transformation of myelodysplastic syndrome to secondary acute myeloid leukemia using a large patient database. Am J Hematol 2012; 87 (09) 853-860
- Swerdlow SH, Campo E, Harris NL. et al.?WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 4th ed.. Lyon, France: IARC; 2017
- Swoboda DM, Sallman DA.?Mutation-driven therapy in MDS. Curr Hematol Malig Rep 2019; 14 (06) 550-560
- Walter MJ, Shen D, Ding L. et al.?Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 2012; 366 (12) 1090-1098
- Bejar R, Stevenson K, Abdel-Wahab O. et al.?Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364 (26) 2496-2506
- Georgiou G, Karali V, Zouvelou C. et al.?Serial determination of FLT3 mutations in myelodysplastic syndrome patients at diagnosis, follow up or acute myeloid leukaemia transformation: incidence and their prognostic significance. Br J Haematol 2006; 134 (03) 302-306
- Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A.?The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000; 96 (12) 3932-3938
- Watanabe-Okochi N, Kitaura J, Ono R. et al.?AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008; 111 (08) 4297-4308
- Lindsley RC, Mar BG, Mazzola E. et al.?Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125 (09) 1367-1376
- Makishima H, Yoshizato T, Yoshida K. et al.?Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet 2017; 49 (02) 204-212
- Ogawa S.?Genetics of MDS. Blood 2019; 133 (10) 1049-1059
- McGowan-Jordan J, Simons A, Schmid M. International Standing Committee on Human Cytogenomic Nomenclature.?ISCN: An International System for Human Cytogenetic Nomenclature (2016). Basel, New York: Karger; 2016: 14
- Chaubey R, Sazawal S, Mahapatra M, Chhikara S, Saxena R.?Does Indian myelodysplastic syndrome have a biology different from that in the West?. Asian Pac J Cancer Prev 2016; 17 (04) 2341-2342
- Lee JJ, Kim HJ, Chung IJ. et al.?Comparisons of prognostic scoring systems for myelodysplastic syndromes: a Korean multicenter study. Leuk Res 1999; 23 (05) 425-432
- Chen B, Zhao WL, Jin J. et al.?Clinical and cytogenetic features of 508 Chinese patients with myelodysplastic syndrome and comparison with those in Western countries. Leukemia 2005; 19 (05) 767-775
- Ma X, Does M, Raza A, Mayne ST.?Myelodysplastic syndromes: incidence and survival in the United States. Cancer 2007; 109 (08) 1536-1542
- Neukirchen J, Schoonen WM, Strupp C. et al.?Incidence and prevalence of myelodysplastic syndromes: data from the D?sseldorf MDS-registry. Leuk Res 2011; 35 (12) 1591-1596
- Algarni AA, Akhtari M, Fu K.?Myelodysplastic syndrome with myelofibrosis transformed to a precursor B-cell acute lymphoblastic leukemia: a case report with review of the literature. Case Rep Hematol 2012; 2012: 207537
- Jiang Y, Eveillard JR, Couturier MA. et al.?Asian population is more prone to develop high-risk myelodysplastic syndrome, concordantly with their propensity to exhibit high-risk cytogenetic aberrations. Cancers (Basel) 2021; 13 (03) 481
- Chaubey R, Sazawal S, Dada R, Mahapatra M, Saxena R.?Cytogenetic profile of Indian patients with de novo myelodysplastic syndromes. Indian J Med Res 2011; 134 (04) 452-457
- Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP.?Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008; 111 (03) 1534-1542
- Haase D, Germing U, Schanz J. et al.?New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 2007; 110 (13) 4385-4395
- Kawata E, Xenocostas A, Hsia CC. et al.?Reevaluating the role of cytogenetic testing in patients with suspected myelodysplastic syndrome in the era of next generation sequencing. Blood 2019; 134 (Suppl. 01) 3438
- Menssen AJ, Walter MJ.?Genetics of progression from MDS to secondary leukemia. Blood 2020; 136 (01) 50-60
- National Comprehensive Cancer Network.?Myelodysplastic syndrome (version 2.2020). Accessed August 10, 2021 at:?http://www.nccn.org/professionals/physician_gls/pdf/mds_blocks.pdf
- Haase D, Stevenson KE, Neuberg D. et al; International Working Group for MDS Molecular Prognostic Committee.?TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019; 33 (07) 1747-1758
- Sanz GF, Iba?ez M, Such E.?Do next-generation sequencing results drive diagnostic and therapeutic decisions in MDS?. Blood Adv 2019; 3 (21) 3454-3460
- Pagliuca S, Gurnari C, Visconte V.?Molecular targeted therapy in myelodysplastic syndromes: new options for tailored treatments. Cancers (Basel) 2021; 13 (04) 784
- Roy S, Coldren C, Karunamurthy A. et al.?Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn 2018; 20 (01) 4-27
- Palomo L, Ib??ez M, Ab?igar M. et al; Spanish Group of MDS (GESMD).?Spanish Guidelines for the use of targeted deep sequencing in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2020; 188 (05) 605-622
- Du MY, Xu M, Deng J. et al.?Evaluation of different scoring systems and gene mutations for the prognosis of myelodysplastic syndrome (MDS) in Chinese population. J Cancer 2020; 11 (02) 508-519
- Platzbecker U. UwePlatzbecker.?Treatment of MDS. Blood 2019; 133 (10) 1096-1107.
Address for correspondence
Publication History
Publication Date:
24 December 2021 (online)
? 2021. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
|?Fig. 1(A)?Complete karyogram shows 45,XY,-7 indicating complete loss of chromosome 7. (B) Metaphase FISH shows orange signal localized to short arm of chromosome 7 (7q31) region and control probe targeting the centromere CEP7 shows green signal. Only one green and orange signal (101G) seen indicates Monosomy 7. (C) Partial karyotype shows interstitial deletion in long arm of chromosome 5. In del(5q), breakpoint occur in q14 and q33. (D) FISH probe localized to short arm of chromosome 5 (5q31) generates orange signal and control probe localized to long arm (5p15.2) and generates green signal. Metaphase FISH shows one orange signal and two green signal (102G) indicating deletion 5q.|
- Tefferi A, Vardiman JW.?Myelodysplastic syndromes. N Engl J Med 2009; 361 (19) 1872-1885
- Devita Jr. VT, Lawrence TA, Rosenberg SA.?DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology. 9th ed.. Philadelphia, PA: Wolters Kluwer; 2011
- Visconte V, Selleri C, Maciejewski JP, Tiu RV.?Molecular pathogenesis of myelodysplastic syndromes. Transl Med UniSa 2014; 8 (04) 19-30
- Steensma DP, Bennett JM.?The myelodysplastic syndromes: diagnosis and treatment. Mayo Clin Proc 2006; 81 (01) 104-130
- Haase D.?Cytogenetic features in myelodysplastic syndromes. Ann Hematol 2008; 87 (07) 515-526
- Shukron O, Vainstein V, K?ndgen A, Germing U, Agur Z.?Analyzing transformation of myelodysplastic syndrome to secondary acute myeloid leukemia using a large patient database. Am J Hematol 2012; 87 (09) 853-860
- Swerdlow SH, Campo E, Harris NL. et al.?WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 4th ed.. Lyon, France: IARC; 2017
- Swoboda DM, Sallman DA.?Mutation-driven therapy in MDS. Curr Hematol Malig Rep 2019; 14 (06) 550-560
- Walter MJ, Shen D, Ding L. et al.?Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 2012; 366 (12) 1090-1098
- Bejar R, Stevenson K, Abdel-Wahab O. et al.?Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364 (26) 2496-2506
- Georgiou G, Karali V, Zouvelou C. et al.?Serial determination of FLT3 mutations in myelodysplastic syndrome patients at diagnosis, follow up or acute myeloid leukaemia transformation: incidence and their prognostic significance. Br J Haematol 2006; 134 (03) 302-306
- Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A.?The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000; 96 (12) 3932-3938
- Watanabe-Okochi N, Kitaura J, Ono R. et al.?AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008; 111 (08) 4297-4308
- Lindsley RC, Mar BG, Mazzola E. et al.?Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125 (09) 1367-1376
- Makishima H, Yoshizato T, Yoshida K. et al.?Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet 2017; 49 (02) 204-212
- Ogawa S.?Genetics of MDS. Blood 2019; 133 (10) 1049-1059
- McGowan-Jordan J, Simons A, Schmid M. International Standing Committee on Human Cytogenomic Nomenclature.?ISCN: An International System for Human Cytogenetic Nomenclature (2016). Basel, New York: Karger; 2016: 14
- Chaubey R, Sazawal S, Mahapatra M, Chhikara S, Saxena R.?Does Indian myelodysplastic syndrome have a biology different from that in the West?. Asian Pac J Cancer Prev 2016; 17 (04) 2341-2342
- Lee JJ, Kim HJ, Chung IJ. et al.?Comparisons of prognostic scoring systems for myelodysplastic syndromes: a Korean multicenter study. Leuk Res 1999; 23 (05) 425-432
- Chen B, Zhao WL, Jin J. et al.?Clinical and cytogenetic features of 508 Chinese patients with myelodysplastic syndrome and comparison with those in Western countries. Leukemia 2005; 19 (05) 767-775
- Ma X, Does M, Raza A, Mayne ST.?Myelodysplastic syndromes: incidence and survival in the United States. Cancer 2007; 109 (08) 1536-1542
- Neukirchen J, Schoonen WM, Strupp C. et al.?Incidence and prevalence of myelodysplastic syndromes: data from the D?sseldorf MDS-registry. Leuk Res 2011; 35 (12) 1591-1596
- Algarni AA, Akhtari M, Fu K.?Myelodysplastic syndrome with myelofibrosis transformed to a precursor B-cell acute lymphoblastic leukemia: a case report with review of the literature. Case Rep Hematol 2012; 2012: 207537
- Jiang Y, Eveillard JR, Couturier MA. et al.?Asian population is more prone to develop high-risk myelodysplastic syndrome, concordantly with their propensity to exhibit high-risk cytogenetic aberrations. Cancers (Basel) 2021; 13 (03) 481
- Chaubey R, Sazawal S, Dada R, Mahapatra M, Saxena R.?Cytogenetic profile of Indian patients with de novo myelodysplastic syndromes. Indian J Med Res 2011; 134 (04) 452-457
- Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP.?Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008; 111 (03) 1534-1542
- Haase D, Germing U, Schanz J. et al.?New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 2007; 110 (13) 4385-4395
- Kawata E, Xenocostas A, Hsia CC. et al.?Reevaluating the role of cytogenetic testing in patients with suspected myelodysplastic syndrome in the era of next generation sequencing. Blood 2019; 134 (Suppl. 01) 3438
- Menssen AJ, Walter MJ.?Genetics of progression from MDS to secondary leukemia. Blood 2020; 136 (01) 50-60
- National Comprehensive Cancer Network.?Myelodysplastic syndrome (version 2.2020). Accessed August 10, 2021 at:?http://www.nccn.org/professionals/physician_gls/pdf/mds_blocks.pdf
- Haase D, Stevenson KE, Neuberg D. et al; International Working Group for MDS Molecular Prognostic Committee.?TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019; 33 (07) 1747-1758
- Sanz GF, Iba?ez M, Such E.?Do next-generation sequencing results drive diagnostic and therapeutic decisions in MDS?. Blood Adv 2019; 3 (21) 3454-3460
- Pagliuca S, Gurnari C, Visconte V.?Molecular targeted therapy in myelodysplastic syndromes: new options for tailored treatments. Cancers (Basel) 2021; 13 (04) 784
- Roy S, Coldren C, Karunamurthy A. et al.?Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn 2018; 20 (01) 4-27
- Palomo L, Ib??ez M, Ab?igar M. et al; Spanish Group of MDS (GESMD).?Spanish Guidelines for the use of targeted deep sequencing in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2020; 188 (05) 605-622
- Du MY, Xu M, Deng J. et al.?Evaluation of different scoring systems and gene mutations for the prognosis of myelodysplastic syndrome (MDS) in Chinese population. J Cancer 2020; 11 (02) 508-519
- Platzbecker U. UwePlatzbecker.?Treatment of MDS. Blood 2019; 133 (10) 1096-1107