 PDF
PDF  Views
Views  Share
Share


Secondary Juvenile Myelo-Monocytic Leukemia in a Patient with Congenital Amegakaryocytic Thrombocytopenia
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(05): 784-786
DOI: DOI: 10.4103/ijmpo.ijmpo_259_19
Isolated thrombocytopenia in childhood is a common phenomenon and mostly occurs due to immune destruction of platelet (immune thrombocytopenic purpura [ITP]). But sometimes, the patient is diagnosed with congenital amegakaryocytic thrombocytopenia (CAMT), a rare inherited bone marrow failure syndrome (IBMFS) affecting a single cell line. The diagnosis and treatment of such a condition is very difficult and long-term follow-up is very important, as these patients may transform into some hematological malignancy. Here, we report one such patient, who was diagnosed with CAMT and on follow-up developed juvenile myelo-monocytic leukemia (JMML), another rare myelodysplastic/myeloproliferative syndrome (MDS/MPN) syndrome.
A 5-year, 9-month-old female child presented to the hematology outpatient department (OPD) in September 2018 with a history of purpuric rash all over the body with gum bleeding for 3 days. There was no history of antecedent fever, jaundice, pain abdomen, shortness of breath, or rash. There was no history of recent exposure to any drug or vaccine. The child was born at full-term normal delivery out of nonconsanguineous marriage. There was no congenital structural abnormality at birth, and no developmental delay was noted. She had one elder sister. On examination, there was no organomegaly, lymphadenopathy, sternal tenderness, or arthropathy. Blood investigations revealed isolated thrombocytopenia with a platelet count of 6000/mm3 on admission. Peripheral blood smear revealed no atypical cells. Lactate dehydrogenase was 420 μ/dl; direct Coombs' test was negative. Other biochemical parameters were normal, and antinuclear antibody was negative. Ultrasonography of the abdomen did not reveal any abnormalities.
The patient was clinically diagnosed as ITP, and was started on prednisolone at 1 mg/kg and intravenous immunoglobulin 1 g/kg for 2 days, along with tranexamic acid. The bleeding manifestation subsided, and platelet count rose to 30,000/mm3 on day 5. The child was discharged with oral medication and followed up in the OPD. However, even after 2 months of steroid therapy, the platelet count did not improve beyond 50,000/mm3.
The patient was planned to be started on 2nd-line therapy, and a bone marrow (BM) study was done in November 2018. BM aspiration revealed cellular trail with normal maturation of erythroid and myeloid lineage without dysplasia [Figure 1a]. However, megakaryocytes were conspicuously absent. No increase in blast percentage was seen. The BM biopsy was normocellular for age, but megakaryocyte was absent [Figure 1b]. No granuloma, infiltration, or fibrosis was seen on biopsy. The features were suggestive of amegakaryocytic thrombocytopenia. The peripheral blood stress cytogenetics did not reveal any chromosomal breakage. The other mutation for IBMFS including thrombopoietin receptor (c-MPL) mutation was not done as the tests were not available in our setup.
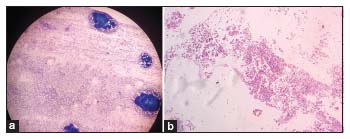
| Figure 1: Initial bone marrow aspiration (a) and biopsy (b) of the patient showing cellular marrow with absent megakaryocyte. The erythroid and myeloid lineage are showing normal maturation without any dysplasia. There was no granuloma, infiltration, or fibrosis. The bone marrow morphology is suggestive of amegakaryocytic thrombocytopenia
The patient's parents were counseled regarding allogeneic BM transplant (BMT). However, no matched sibling donor was available. The parents were not willing for haploidentical transplant, so a matched unrelated donor (MUD) search was initiated. In the meantime, she was started on anti thymocyte globulin + cyclosporin and eltrombopag in January 2019. However, she continued to have persistent thrombocytopenia in the range of 15,000/mm3 to 65,000/mm3, with occasional purpuric spots and gum bleeding and occasional platelet transfusion support. Due to financial issues, she stopped eltrombopag after 5 months of treatment and continued only oral cyclosporine.
In October 2019, after 1 year, 1 month of diagnosis, she was again admitted with fever for 5 days, gum bleeding, and excessive weakness. There was also pain in the bilateral ankle joints. On examination, there was pallor; bilateral cervical lymph nodes were measuring 0.5 cm × 0.5 cm; there was mobile, nontender, per abdomen; liver was of 2 cm; spleen was of 4 cm; there was no ascites; and chest revealed bilateral crepitation. The blood report showed hemoglobin of 3.7 g/dl, total leukocyte count of 45,000/mm3, and platelet count of 10,000/mm3. The differential count showed neutrophil of 70%, lymphocyte of 6%, monocyte of 5%, eosinophil of 1%, myelocyte of 14%, metamyelocyte of 4%, and nucleated RBC of 4 per 100 WBC counted. Peripheral smear showed the presence of microcytic hypochromic anemia and polychromasia with occasional nRBCs. The WBC showed dysplastic hypogranular neutrophils, pseudo Pelger–Huet anomaly, and bilobed neutrophils [Figure 2a]. The repeat BM aspiration and biopsy revealed cellular marrow with dysplasia noted in all cell lineages and increased blast of 14% [Figure 2b]. Flow cytometry confirmed the presence of 16% myeloblasts, with increase in monocytic population. The cytogenetic study revealed normal female karyotype. The patient was diagnosed as amegakaryocytic thrombocytopenia transformed into JMML. The patient is planned to be treated with intensive chemotherapy using Fludarabine, Arabinoside C, GCSF- Idarubicine regimen followed by an immediate Haplo transplant from her sister who is 6/10 matched.
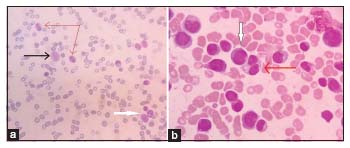
| Figure 2: Repeat peripheral smear (a) and bone marrow aspiration (b) suggestive of juvenile myelo monocytic leukemia. The peripheral smear (a) showing the presence of dysplastic hypogranular hypolobated neutrophils with pseudo Pelger–Huet anomaly (red arrow), monocyte (black arrow), and myeloblast (white arrow). The bone marrow aspiration (b) smear is showing the presence of myeloblast (white arrow) and dysplasia in myeloid lineage (red arrow). The features are suggestive of juvenile myelo monocytic leukemia
CAMT is an autosomal recessive IBMFS which occurs due to congenital absence or deficiency of cMPL, the receptor of thrombopoietin, resulting in reduced or absent megakaryocyte proliferation and maturation. The suspicion arises when children with isolated thrombocytopenia present with excessive bleeding manifestation and do not respond well with conventional treatment for ITP.[1] The diagnosis is confirmed by documenting the absence or reduced number of megakaryocytes in the BM.[2] A germline cMPL mutation can conclusively prove the diagnosis, but most centers do not have such facilities.[3],[4] Inherited thrombocytopenia in children may be found in other IBMFS also. Hence, thorough clinical examination, for characteristic phenotypic abnormalities (e.g. thrombocytopenia-absent radius syndrome) and stress cytogenetics to test chromosomal hypersensitivity to DNA cross-linking agents (to rule out early Fanconi's anemia), should be done before confirming CAMT.[2]
Currently, there are only limited data available on the clinical course and the outcome of patients with CAMT. The only curative treatment is allogeneic BMT from a matched sibling or unrelated donor, if available.[5] The other therapies including immunosuppressor therapies or thrombopoietin mimetic have been tried in different inherited thrombocytopenias with limited success.[6],[7] Because our patient did not have human leucocyte antigen-matched donor, a possible MUD search was started.
The increased propensity to develop different malignancies, including hematological malignancies, is a well-known phenomenon for different IBMFS conditions. The alteration of genetic landscape with acquiring new mutations and defect in DNA repair mechanism leads to development of myelodysplasia and leukemia in such patients. Little is known about long changes in CAMT patients. Although the risk for malignant transformation seems to be very low in contrast to that observed in other BM failure syndromes, the scarcity of literature and rarity of the disease make it difficult to study the long-term outcome of CAMT patients.[8] Alter described two CAMT patients: one developed acute myeloid leukemia (AML) at the age of 16 years and another developed myelodysplasia with abnormalities of chromosome 19.[9] Another study of long-term follow-up of CAMT patients showed the development of myelodysplasia in one out of twenty patients, who progressed to develop AML in spite of undergoing allogeneic BMT.[1] Our patient also developed JMML, a rare MDS/MPN overlap syndrome, hitherto not mentioned in previous literature. The diagnosis was confirmed by morphological and flow cytometry analysis, though conventional karyotyping did not reveal any chromosomal alteration.
CAMT is always a difficult-to-diagnose and difficult-to-treat disorder. Although allogeneic BMT is the only curative option, it cannot prevent further genetic alteration and possible development of second malignancy. Increased vigilance and long-term follow-up is required for such patients.
Publication History
Received: 19 December 2019
Accepted: 28 April 2020
Article published online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Isolated thrombocytopenia in childhood is a common phenomenon and mostly occurs due to immune destruction of platelet (immune thrombocytopenic purpura [ITP]). But sometimes, the patient is diagnosed with congenital amegakaryocytic thrombocytopenia (CAMT), a rare inherited bone marrow failure syndrome (IBMFS) affecting a single cell line. The diagnosis and treatment of such a condition is very difficult and long-term follow-up is very important, as these patients may transform into some hematological malignancy. Here, we report one such patient, who was diagnosed with CAMT and on follow-up developed juvenile myelo-monocytic leukemia (JMML), another rare myelodysplastic/myeloproliferative syndrome (MDS/MPN) syndrome.
A 5-year, 9-month-old female child presented to the hematology outpatient department (OPD) in September 2018 with a history of purpuric rash all over the body with gum bleeding for 3 days. There was no history of antecedent fever, jaundice, pain abdomen, shortness of breath, or rash. There was no history of recent exposure to any drug or vaccine. The child was born at full-term normal delivery out of nonconsanguineous marriage. There was no congenital structural abnormality at birth, and no developmental delay was noted. She had one elder sister. On examination, there was no organomegaly, lymphadenopathy, sternal tenderness, or arthropathy. Blood investigations revealed isolated thrombocytopenia with a platelet count of 6000/mm3 on admission. Peripheral blood smear revealed no atypical cells. Lactate dehydrogenase was 420 μ/dl; direct Coombs' test was negative. Other biochemical parameters were normal, and antinuclear antibody was negative. Ultrasonography of the abdomen did not reveal any abnormalities.
The patient was clinically diagnosed as ITP, and was started on prednisolone at 1 mg/kg and intravenous immunoglobulin 1 g/kg for 2 days, along with tranexamic acid. The bleeding manifestation subsided, and platelet count rose to 30,000/mm3 on day 5. The child was discharged with oral medication and followed up in the OPD. However, even after 2 months of steroid therapy, the platelet count did not improve beyond 50,000/mm3.
The patient was planned to be started on 2nd-line therapy, and a bone marrow (BM) study was done in November 2018. BM aspiration revealed cellular trail with normal maturation of erythroid and myeloid lineage without dysplasia [Figure 1a]. However, megakaryocytes were conspicuously absent. No increase in blast percentage was seen. The BM biopsy was normocellular for age, but megakaryocyte was absent [Figure 1b]. No granuloma, infiltration, or fibrosis was seen on biopsy. The features were suggestive of amegakaryocytic thrombocytopenia. The peripheral blood stress cytogenetics did not reveal any chromosomal breakage. The other mutation for IBMFS including thrombopoietin receptor (c-MPL) mutation was not done as the tests were not available in our setup.
| Figure 1: Initial bone marrow aspiration (a) and biopsy (b) of the patient showing cellular marrow with absent megakaryocyte. The erythroid and myeloid lineage are showing normal maturation without any dysplasia. There was no granuloma, infiltration, or fibrosis. The bone marrow morphology is suggestive of amegakaryocytic thrombocytopenia
The patient's parents were counseled regarding allogeneic BM transplant (BMT). However, no matched sibling donor was available. The parents were not willing for haploidentical transplant, so a matched unrelated donor (MUD) search was initiated. In the meantime, she was started on anti thymocyte globulin + cyclosporin and eltrombopag in January 2019. However, she continued to have persistent thrombocytopenia in the range of 15,000/mm3 to 65,000/mm3, with occasional purpuric spots and gum bleeding and occasional platelet transfusion support. Due to financial issues, she stopped eltrombopag after 5 months of treatment and continued only oral cyclosporine.
In October 2019, after 1 year, 1 month of diagnosis, she was again admitted with fever for 5 days, gum bleeding, and excessive weakness. There was also pain in the bilateral ankle joints. On examination, there was pallor; bilateral cervical lymph nodes were measuring 0.5 cm × 0.5 cm; there was mobile, nontender, per abdomen; liver was of 2 cm; spleen was of 4 cm; there was no ascites; and chest revealed bilateral crepitation. The blood report showed hemoglobin of 3.7 g/dl, total leukocyte count of 45,000/mm3, and platelet count of 10,000/mm3. The differential count showed neutrophil of 70%, lymphocyte of 6%, monocyte of 5%, eosinophil of 1%, myelocyte of 14%, metamyelocyte of 4%, and nucleated RBC of 4 per 100 WBC counted. Peripheral smear showed the presence of microcytic hypochromic anemia and polychromasia with occasional nRBCs. The WBC showed dysplastic hypogranular neutrophils, pseudo Pelger–Huet anomaly, and bilobed neutrophils [Figure 2a]. The repeat BM aspiration and biopsy revealed cellular marrow with dysplasia noted in all cell lineages and increased blast of 14% [Figure 2b]. Flow cytometry confirmed the presence of 16% myeloblasts, with increase in monocytic population. The cytogenetic study revealed normal female karyotype. The patient was diagnosed as amegakaryocytic thrombocytopenia transformed into JMML. The patient is planned to be treated with intensive chemotherapy using Fludarabine, Arabinoside C, GCSF- Idarubicine regimen followed by an immediate Haplo transplant from her sister who is 6/10 matched.
| Figure 2: Repeat peripheral smear (a) and bone marrow aspiration (b) suggestive of juvenile myelo monocytic leukemia. The peripheral smear (a) showing the presence of dysplastic hypogranular hypolobated neutrophils with pseudo Pelger–Huet anomaly (red arrow), monocyte (black arrow), and myeloblast (white arrow). The bone marrow aspiration (b) smear is showing the presence of myeloblast (white arrow) and dysplasia in myeloid lineage (red arrow). The features are suggestive of juvenile myelo monocytic leukemia
CAMT is an autosomal recessive IBMFS which occurs due to congenital absence or deficiency of cMPL, the receptor of thrombopoietin, resulting in reduced or absent megakaryocyte proliferation and maturation. The suspicion arises when children with isolated thrombocytopenia present with excessive bleeding manifestation and do not respond well with conventional treatment for ITP.[1] The diagnosis is confirmed by documenting the absence or reduced number of megakaryocytes in the BM.[2] A germline cMPL mutation can conclusively prove the diagnosis, but most centers do not have such facilities.[3],[4] Inherited thrombocytopenia in children may be found in other IBMFS also. Hence, thorough clinical examination, for characteristic phenotypic abnormalities (e.g. thrombocytopenia-absent radius syndrome) and stress cytogenetics to test chromosomal hypersensitivity to DNA cross-linking agents (to rule out early Fanconi's anemia), should be done before confirming CAMT.[2]
Currently, there are only limited data available on the clinical course and the outcome of patients with CAMT. The only curative treatment is allogeneic BMT from a matched sibling or unrelated donor, if available.[5] The other therapies including immunosuppressor therapies or thrombopoietin mimetic have been tried in different inherited thrombocytopenias with limited success.[6],[7] Because our patient did not have human leucocyte antigen-matched donor, a possible MUD search was started.
The increased propensity to develop different malignancies, including hematological malignancies, is a well-known phenomenon for different IBMFS conditions. The alteration of genetic landscape with acquiring new mutations and defect in DNA repair mechanism leads to development of myelodysplasia and leukemia in such patients. Little is known about long changes in CAMT patients. Although the risk for malignant transformation seems to be very low in contrast to that observed in other BM failure syndromes, the scarcity of literature and rarity of the disease make it difficult to study the long-term outcome of CAMT patients.[8] Alter described two CAMT patients: one developed acute myeloid leukemia (AML) at the age of 16 years and another developed myelodysplasia with abnormalities of chromosome 19.[9] Another study of long-term follow-up of CAMT patients showed the development of myelodysplasia in one out of twenty patients, who progressed to develop AML in spite of undergoing allogeneic BMT.[1] Our patient also developed JMML, a rare MDS/MPN overlap syndrome, hitherto not mentioned in previous literature. The diagnosis was confirmed by morphological and flow cytometry analysis, though conventional karyotyping did not reveal any chromosomal alteration.
CAMT is always a difficult-to-diagnose and difficult-to-treat disorder. Although allogeneic BMT is the only curative option, it cannot prevent further genetic alteration and possible development of second malignancy. Increased vigilance and long-term follow-up is required for such patients.
Conflict of Interest
There are no conflicts of interest.
References
- King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: A retrospective clinical analysis of 20 patients. Br J Haematol 2005; 131: 636-44
- Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol 2009; 146: 3-16
- Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A. et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001; 97: 139-46
- Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA. et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proceed Natl Academy Sci United States Am 1999; 96: 3132-36
- Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S. et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001; 97: 139-46
- Zaninetti C, Gresele P, Bertomoro A. et al. Eltrombopag for the treatment of inherited thrombocytopenias: a phase 2 clinical trial. Haematologica 2020; 105: 820-8
- Pecci A. Pathogenesis and management of inherited thrombocytopenias: Rationale for the use of thrombopoietin-receptor agonists. Int J Hematol 2013; 98: 34-47
- Maserati E, Panarello C, Morerio C, Valli R, Pressato B, Patitucci F. et al. Clonal chromosome anomalies and propensity to myeloid malignancies in congenital amegakaryocytic thrombocytopenia (OMIM 604498). Haematologica 2008; 93: 1271-3
- Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Stranahan RA, Orkin SH, Look AT, Ginsburg D. editors Nathan and Oski's Hematology of Infancy and Childhood. Philadelphia: Saunders W.B; 2003: 280-365
Address for correspondence
Publication History
Received: 19 December 2019
Accepted: 28 April 2020
Article published online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: Initial bone marrow aspiration (a) and biopsy (b) of the patient showing cellular marrow with absent megakaryocyte. The erythroid and myeloid lineage are showing normal maturation without any dysplasia. There was no granuloma, infiltration, or fibrosis. The bone marrow morphology is suggestive of amegakaryocytic thrombocytopenia
| Figure 2: Repeat peripheral smear (a) and bone marrow aspiration (b) suggestive of juvenile myelo monocytic leukemia. The peripheral smear (a) showing the presence of dysplastic hypogranular hypolobated neutrophils with pseudo Pelger–Huet anomaly (red arrow), monocyte (black arrow), and myeloblast (white arrow). The bone marrow aspiration (b) smear is showing the presence of myeloblast (white arrow) and dysplasia in myeloid lineage (red arrow). The features are suggestive of juvenile myelo monocytic leukemia
References
- King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: A retrospective clinical analysis of 20 patients. Br J Haematol 2005; 131: 636-44
- Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol 2009; 146: 3-16
- Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A. et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001; 97: 139-46
- Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA. et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proceed Natl Academy Sci United States Am 1999; 96: 3132-36
- Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S. et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001; 97: 139-46
- Zaninetti C, Gresele P, Bertomoro A. et al. Eltrombopag for the treatment of inherited thrombocytopenias: a phase 2 clinical trial. Haematologica 2020; 105: 820-8
- Pecci A. Pathogenesis and management of inherited thrombocytopenias: Rationale for the use of thrombopoietin-receptor agonists. Int J Hematol 2013; 98: 34-47
- Maserati E, Panarello C, Morerio C, Valli R, Pressato B, Patitucci F. et al. Clonal chromosome anomalies and propensity to myeloid malignancies in congenital amegakaryocytic thrombocytopenia (OMIM 604498). Haematologica 2008; 93: 1271-3
- Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Stranahan RA, Orkin SH, Look AT, Ginsburg D. editors Nathan and Oski's Hematology of Infancy and Childhood. Philadelphia: Saunders W.B; 2003: 280-365