 PDF
PDF  Views
Views  Share
Share


Role of Conventional Cytogenetics and FISH in the Laboratory Work Up of Plasma Cell Dyscrasias
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(05): 505-514
DOI: DOI: 10.1055/s-0043-1762920
Abstract
Plasma cell dyscrasias are a heterogeneous group of neoplasms characterized by abnormal proliferation of plasma cells with or without over production of monoclonal immunoglobulins. Chromosomal abnormalities are acquired either early in the course of the disease or during disease progression. Plasma cell dyscrasias are categorized into multiple cytogenetic subtypes that form an integral component of risk-stratified treatment protocols. The primary genetic events are IgH gene translocations and non-random gains of chromosomes 3/5/7/9/11/15/19 and or 21. The secondary genetic events consist of chromosome 1 abnormalities (1p deletion and 1q gain or amplification), deletion 17p/TP53, deletion 13q, and MYC gene rearrangements. Plasma cells being at the end of differentiation spectrum of B cells, have low proliferative potential precluding the use of karyotyping in identification of chromosomal abnormalities. Analysis of enriched plasma cells using interphase fluorescent in situ hybridization (FISH) is the technique of choice for identifying these abnormalities. It is essential to enrich plasma cells before the FISH analysis, and numerous plasma cell enrichment techniques have been described. In the paper, we review the cytogenetic approach to identify clinically significant genetic aberrations including the effective use of FISH panels and plasma cell enrichment techniques.
Keywords
plasma cell dyscrasia - multiple myeloma - FISH - MGUS - SMM - plasma cell leukemia - cytogenetics in myelomaAuthors' Contributions
A.D. and M.P. wrote the original draft, S.D. prepared the figures; M.P. and A.D. have full access to all data and the final responsibility for publication. All authors reviewed the manuscript draft submitted for publication.
Publication History
Article published online:
24 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Plasma cell dyscrasias are a heterogeneous group of neoplasms characterized by abnormal proliferation of plasma cells with or without over production of monoclonal immunoglobulins. Chromosomal abnormalities are acquired either early in the course of the disease or during disease progression. Plasma cell dyscrasias are categorized into multiple cytogenetic subtypes that form an integral component of risk-stratified treatment protocols. The primary genetic events are IgH gene translocations and non-random gains of chromosomes 3/5/7/9/11/15/19 and or 21. The secondary genetic events consist of chromosome 1 abnormalities (1p deletion and 1q gain or amplification), deletion 17p/TP53, deletion 13q, and MYC gene rearrangements. Plasma cells being at the end of differentiation spectrum of B cells, have low proliferative potential precluding the use of karyotyping in identification of chromosomal abnormalities. Analysis of enriched plasma cells using interphase fluorescent in situ hybridization (FISH) is the technique of choice for identifying these abnormalities. It is essential to enrich plasma cells before the FISH analysis, and numerous plasma cell enrichment techniques have been described. In the paper, we review the cytogenetic approach to identify clinically significant genetic aberrations including the effective use of FISH panels and plasma cell enrichment techniques.
Keywords
plasma cell dyscrasia - multiple myeloma - FISH - MGUS - SMM - plasma cell leukemia - cytogenetics in myelomaIntroduction
Plasma cell dyscrasias are a group of heterogeneous neoplasms characterized by clonal proliferation of plasma cells. Based on the percentage of plasma cell infiltration in the bone marrow, the type of monoclonal protein, association of CRAB (hypercalcemia, renal impairment, anemia, or lytic bone lesions) plasma cell dyscrasias are classified as[1] [2]:
IgM monoclonal gammopathy of undetermined significance
Non IgM monoclonal gammopathy of undetermined significance
Smoldering (asymptomatic) plasma cell myeloma
Multiple myeloma (plasma cell myeloma):
➢ Multiple myeloma NOS
➢ Multiple myeloma with recurrent genetic abnormality
➢ Multiple myeloma with CCND family translocation
➢ Multiple myeloma with MAF family translocation
➢ Multiple myeloma with NSD2 family translocation
➢ Multiple myeloma with hyperdiploidy
Solitary plasmacytoma of bone
Extraosseous plasmacytoma
Monoclonal immunoglobulin deposition disease
Immunoglobulin light chain amyloidosis (AL)
Localized AL amyloidosis
Light chain and heavy chain deposition disease
The plasma cells undergo several rounds of differentiation in the bone marrow and secondary lymphoid organs and involve V(D)J rearrangements and somatic hypermutation with class switch recombination.[3] Sentinel chromosomal abnormalities acquired in the process of maturation and differentiation result in neoplastic transformation of the plasma cells.[3] These cytogenetic abnormalities are integral to risk stratified treatment protocols. The founding or primary chromosomal abnormalities that occur early in the course of the disease are IgH gene rearrangements and aneuploidy.[3] [4] Secondary chromosomal abnormalities are either acquired or enriched at disease progression and include deletion of short arm of chromosome 17 (deletion 17p/TP53), deletion of short arm of chromosome 1 (1p deletion), gain or amplification of long arm of chromosome 1 (1q gain/amp), deletion of long arm of chromosome 13 (13q deletion) and MYC gene rearrangements.[4]
In IgH gene rearrangements, the promoter sequences of the IgH enhancers cause overexpression of the partner genes. The recurrent IgH translocations in multiple myeloma (MM), involve CCND1 at 11q13, CCND3 at 6p21, FGFR3/MMSET/CCND2 at 4p16 resulting in increased expression of cyclin D family of genes that promote cell cycle progression and increased proliferation. IgH translocations involving the MAF family of genes include MAF at 16q23 and MAFB at 20q11, resulting in the upregulation of MAF-associated transcription process.[5]
Aneuploidies include hyperdiploid MM and the non-hyperdiploid MM. Hyperdiploidy in plasma cells neoplasms is characterized by nonrandom gains of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21. The gains of chromosomes result in gene dosage effects, altering gene expression. Gains of chromosome 11 is associated with the overexpression of the CCND1 gene. Nonhyperdiploid MM includes hypodiploid (<45 href="https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0043-1762920#JR222450997-5" xss=removed>5] [6]
Secondary cytogenetic abnormalities can be present either at diagnosis or may be enriched or acquired during progression of the disease. The molecular mechanisms that promote progression include the activation of the RAS pathway and MYC overexpression accompanied by DNA hypomethylation leading to genomic instability ([Fig. 1]).
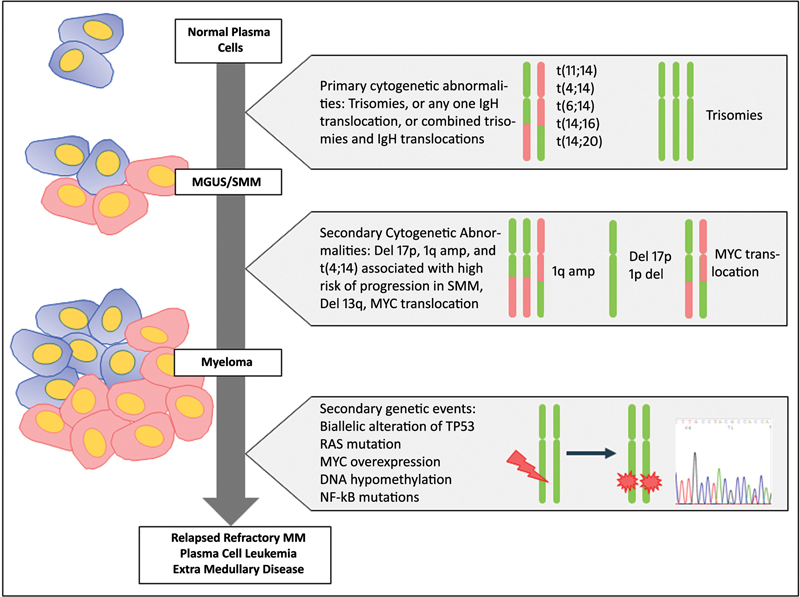
| Fig 1 :Cytogenetic and genetic abnormalities occurring in the evolution of plasma cell dyscrasias.
In this review, we will focus on the role of cytogenetics for the work up of plasma cell dyscrasias.
Cytogenetic Risk Stratification
MM is a heterogeneous disease characterized by multiple genetic subtypes that have varied response to treatment and are an integral component of risk stratification of the disease. The impact of cytogenetic abnormalities on time to progression (TTP) varies according to the type of plasma cell dyscrasia. In MM, the standard risk abnormalities include t(11;14), t(6;14), and hyperdiploidy (gains of 3/5/7/9/11/15/19 and or 21) and high-risk abnormalities include t(4;14), t(14;16), t(14;20), deletion of short arm of chromosome 17 (Del 17p), gain or amplification of long arm of chromosome 1 (1q gain/amp), deletion of short arm of chromosome 1 (1p deletion), MYC gene rearrangements, and deletion of long arm of chromosome 13 (13q deletion).[1] [2] [7] [8]
The revised international staging system for myeloma established by the International Myeloma Working Group includes high-risk cytogenetic abnormalities such as t(4;14), t(14;16) deletion 17p/TP53 along with serum albumin, serum β-2 microglobulin, and serum lactate dehydrogenase levels. The second revision of the RISS excluded t(14;16) and included 1q amplification in the scoring system.[9]
The Mayo Clinic includes both the standard risk and high-risk cytogenetic abnormalities in the Stratification for Myeloma and Risk Adapted Therapy (mSMART) protocol ([Table 1]).[10] [11] [12] [13]
Triple-hit myeloma: ≥3 high-risk abnormalities |
a Trisomies may ameliorate.
b By FISH or equivalent method.
c Cutoffs vary.
d t(11;14) may be associated with plasma cell leukemia.
Double-Hit and Triple-Hit Myeloma
The mSMART 3.0 proposed by the Mayo Clinic has proposed a concept of double-hit myeloma and triple-hit myeloma similar to the concept seen in lymphomas.[14] These are defined as follows:
High-risk abnormalities include deletion 17p/TP53, 1q gain/amp, t(4;14), t(14;16), t(14;20)
Double-hit MM (DH-MM) includes any two high-risk abnormalities
Triple-hit MM (TH-MM) includes three or more high-risk abnormalities
These are considered as the 'ultra-high risk' abnormalities showing rapid disease progression and shorter overall survival as compared to MM having a single or no high-risk abnormality. The most common high-risk abnormalities seen were 1q gain/amp, t(4;14), deletion 17p/TP53 in several studies.[15] [16]
Cytogenetic Lab Approach in MM
The cytogenetic strategy in plasma cell dyscrasia is based predominantly on FISH analysis of neoplastic plasma cells. The use of karyotyping has decreased in recent years due to reasons explained below. Although single nucleotide polymorphism (SNP) array has been used to identify copy number abnormalities, it does not identify fusions and the results have to be integrated with FISH analysis.[17] [18]
Transcriptomic analysis has been used to identify high-risk gene expression signatures.[19] [20] This review will focus on FISH-based approach to identify the genetic subtypes.
Karyotyping (Conventional Cytogenetics)
Karyotyping relies on the ability of plasma cells to divide, which is limited, making it difficult to acquire metaphases for study. Stimulants such as lipopolysaccharide (LPS), 12-O-tetradecanoylphorbol-13-acetate (TPA or phorbol 12-myristate 13-acetate), and cytokines (interleukin-6 and granulocyte–macrophage colony-stimulating are used to increase the yield of metaphases, their role in IgM-negative B cells being limited.[21] [22] Oligonucleotides containing CpG motif, such as synthetic DSP30, can stimulate cells of the immune system in vitro and hence can be used to increase the yield of metaphases in vitro.[23] Various cytokines such as IL-10, IL-2, and TNF-alpha can also be used as mitogens for increasing the yield of metaphases.[23] Though G-banded analysis has the advantage of whole genome analysis at a low resolution ([Fig. 2A]), low or no yield of metaphases from the plasma cells limits its utility in identifying the subtypes. Karyotyping fails to identify cryptic translocations involving the IgH locus such as t(4;14) and t(14;16) and a subset of cryptic 17p deletions. Hence, FISH is a superior tool with better sensitivity and specificity in identifying the clinically relevant genetic subtypes.[17] [24]
mSMART 3.0
(risk stratification of active MM)
Standard risk
Trisomies of 3/5/7/9/11/15/19 and or 21*
t(4;14)
t(11;14) [d]
t(14;16)
t(6;14)
t(14;20)
Del 17p
TP53 mutation
1q gain
• R-ISS stage 3
• High-plasma cell S-phase[c]
• GEP: High-risk signature
Double-hit myeloma: any 2 high-risk abnormalities
Triple-hit myeloma: ≥3 high-risk abnormalities

| Figure 2:(A) GTG banded karyotype image showing hyperdiploidy with deletion of long arm of chromosome 6 and a balanced translocation between the long arms of chromosome of 8 and 22. (B) Interphase FISH with IgH break-apart probe positive for IgH rearrangement. (C) Interphase FISH with CCND1::IgH dual color dual fusion probe positive for t(11;14). (D) Interphase FISH with LSI D5S23, D5S721/CEP 9/CEP 15 tricolor probe depicting trisomies for chromosomes 5,9,15. (E) Interphase FISH with 1p(CDKN2C)/1q(CKS1B) LSI probe depicting deletion of 1p(CDKN2C) and gain of 1q(CKS1B). (F) Interphase FISH targetingTP53 gene depicting TP53 deletion.
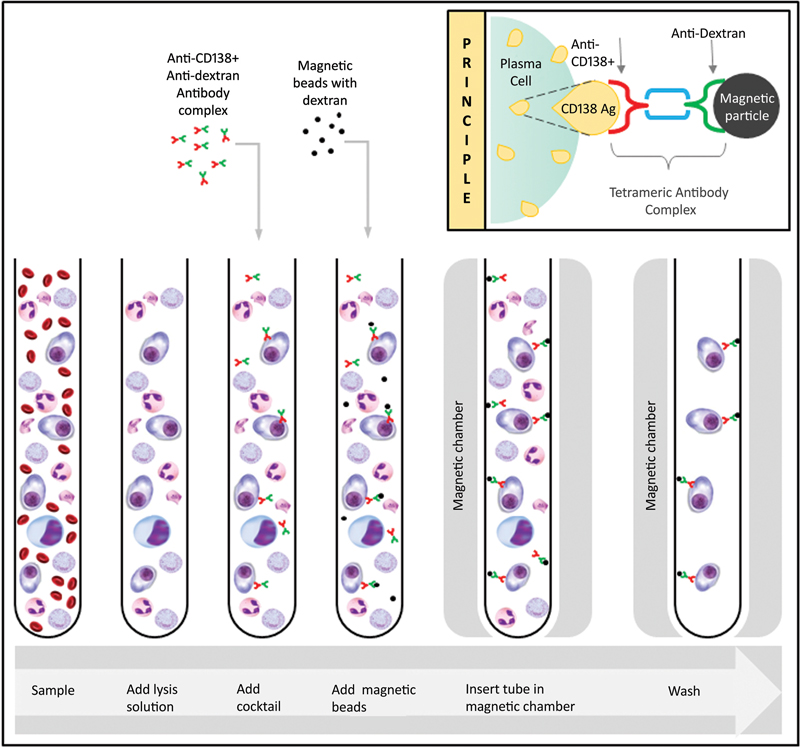
| Figure 3: Magnetic cell sorting (MACS) of CD138+ plasma cells using anti-CD138 and anti-dextran complex through positive selection of plasma cells in a magnetic chamber.
Fluorescent in-situ Hybridization
FISH does not require live cells and can be performed on interphase cells. Plasma cell infiltration of the bone marrow can be patchy in MM, and the percentage of plasma cells in the marrow may vary. It is essential to enrich plasma cells before application of probes and performing the FISH analysis. Currently, FISH is the preferred tool to identify the genetic subtypes for risk stratification in MM patients.[24] [25] [26] [27]
Various Plasma Cell Enrichment Techniques
1) Magnetic cell sorting (MACS)
2) Fluorescence activating cell sorting (FACS)
3) Targeted manual sorting
4) Customized automated image analysis
5) Cytoplasmic immunoglobulin FISH (cIg-FISH)
6) Target FISH[28]
Magnetic Cell Sorting
The adhesion of the plasma cells to an antibody cocktail serves as a basic principle for sorting the cells. Fresh heparin bone marrow samples are treated with an anti-CD138 and anti-dextran antibody complex. Magnetic beads are added that bind to the dextran complex and this is subjected to a magnetic chamber for separation. The unbound particles are washed off and the mixture obtained contains the sorted plasma cells ([Fig. 3]). The technique is cost-effective and requires fresh samples, as the yield decreases with the age of the sample.[28]
Fluorescence Activating Cell Sorting
Flow cytometric sorting of neoplastic plasma cells is performed using a cocktail of various antibodies (e.g., anti-CD45, anti-CD56, and anti-CD38) along with assessment of light scattering ability of plasma cells. FACS is more efficient than MACS as it uses multiple antibodies and parameters to identify and sort neoplastic plasma cells, thereby increasing the yield of plasma cells available for characterization. Higher capital and maintenance costs and restricted access coupled with cost of monoclonal antibodies have limited the use of sorter in most of the routine diagnostic laboratories.[28] [29]
Targeted Manual Sorting
Targeted manual sorting relies on morphological identification of large mononuclear cells. One of the limitations of Target FISH is the intensity of signals that is weak and not uniform across the slide. The technique is subjective, requires skilled manpower, is time consuming with an increase possibility of false-negative results.[28]
Customized Automated Image Analysis
FISH slides are subjected to automated slide scanning where the mono nuclear cells are identified using a software classifier. The ability to count more cells increases the sensitivity of this technique as compared to manual analysis. The major drawback is the capital costs and infrastructure required to store and analyze the images.[28]
Cytoplasmic Immunoglobulin
Plasma cells are differentiated from other cells in the marrow by staining them with anti-kappa or anti-lambda antibodies (cytoplasmic immunoglobulin FISH [clg-FISH]). The method is tedious, requires additional time for careful meticulous examination of the slides. In cases with aggregation of small plasma cells, the analysis is difficult. The modified cIg FISH has refined the identification of plasma cells by fixing the plasma cells to avoid clumping or aggregation by replacing 96-% ethanol wash with 100-% methanol. The method is the preferred technique of FISH on plasma cells in a large number of laboratories.[30] [31] [32]
Target FISH
Plasma cells are sorted by centrifugation using Ficoll and are then stained with May–Grünwald Giemsa (MGG) stain. Plasma cells are identified based on morphology and are captured using an automated system. These slides are then de-stained and FISH is performed on the same slides. Plasma cells are identified based on the analysis of previously captured and analyzed images and FISH signals are recorded on the same cell. The disadvantage is that thousands of images are captured and there are reports of discordant results between the images captured.[33]
Quantitative Multigene Fluorescent in-situ Hybridization
Quantitative multigene fluorescent in-situ hybridization (QM-FISH) is a cytological high-resolution technique used to identify heterogeneity and clonal evolution in cases of MM. Single cell analysis could provide new insights into the clonal evolution in MM. Because MM is a heterogenous disease, clonal evolution is a continuous process and QM-FISH can only identify selective abnormalities. QM-FISH could prove to be an important tool to analyze the new emerging clones with its sensitivity being similar to conventional FISH.[34]
Pre-analytical Variables and Quality Control
The first pull bone marrow aspirate sample is the preferred sample for cytogenetic studies in MM. The sample should be transported as soon as possible to the laboratory and processed with minimum delay. A delay in the transport and processing of samples results in the depletion of the plasma cells impacting the results of the study. Hemolyzed and clotted samples adversely impact the plasma cell enrichment process and a repeat sample should be requested. While standardizing the plasma cell enrichment process using MACS, flow cytometry-based analysis should be performed to evaluate the efficiency of the enrichment process to positively select the CD138-positive plasma cells. The laboratory should perform analysis on known positive and negative samples to calculate the cut-off for each probe. A recommendation of 10-% for fusion or break-apart probes and 20-% for numerical abnormalities has been mentioned in some studies.[35]
These recommendations are not universally accepted and ideally the laboratories should define their own cut-off values for each probe. It is expected that in samples with plasma cell enrichment the primary abnormalities will be present in the majority of the cells as compared to the secondary abnormalities. Ideally, 100 cells should be evaluated by at least two analysts.[35] Cut-off values for each probe can be calculated by either using CV with standard deviation and beta inverse function or using the Excel (Microsoft, Redmond, WA) statistical function CRITBINOM (n, p, α) with a confidence level of 95-%.[36]
FISH studies on bone marrow biopsies are technically challenging with high frequency of failure due to the use of acids in decalcification process. Fixation in 10-% neutral buffered formalin for at least 24 hours and EDTA-based decalcification are recommended for optimal FISH analysis on bone marrow biopsy specimens. In patients, showing patchy interstitial infiltration by plasma cells in the bone marrow biopsy, identifying the plasma cells is challenging and essential expertise is required to avoid false-negative results.
Probe Selection
To identify deletions/amplifications dual-color locus specific probes with an internal control is recommended. A break-apart probe is used for identifying IgH and MYC rearrangements. Dual color fusion probes are used to identify specific IgH partners.[35] [37]
Step Wise FISH Strategy
FISH analysis to identify the clinically relevant genetic subtypes is performed in a stepwise manner to judiciously utilize the enriched plasma cells and save costs ([Table 2]). The first step involves testing for 17p,13q deletion, 1q gain/1p deletion, IgH and MYC rearrangement using respective break-apart probes and centromeric probes targeting 5,9,15 to identify trisomies. Based on the results of the initial panel, patient samples positive for IgH rearrangement ([Fig. 2B]) are reflex tested using specific fusion probes to identify the partner. Follow-up samples are tested for deletion 17p, MYC rearrangements, and 1q gain/amp.
|
At diagnosis |
|---|
|
IgH gene rearrangement |
|
Deletion 17p |
|
1q gain/amplification |
|
1p deletion |
|
8q24.1 rearrangement, MYC break apart |
|
Deletion 13q |
|
CEP 5/9/15 (By FISH or flow ploidy) |
|
If positive for IGH gene rearrangement |
|
t(11;14)(q13;q32), CCND1 IgH fusion |
|
t(6;14)(p21;q32) CCND3::IgH fusion |
|
t(4;14)(p16.3;q32) FGFR3::IgH fusion |
|
t(14;16)(q32;q23) IgH::MAF fusion |
|
t(14;20)(q32;q12) IgH::MAFB fusion |
|
At follow-up |
|
Deletion 17p |
|
1q gain/amplification |
|
8q24.1 rearrangement, MYC break apart |
Address for correspondence
Publication History
Article published online:
24 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig 1 :Cytogenetic and genetic abnormalities occurring in the evolution of plasma cell dyscrasias.
| Figure 2:(A) GTG banded karyotype image showing hyperdiploidy with deletion of long arm of chromosome 6 and a balanced translocation between the long arms of chromosome of 8 and 22. (B) Interphase FISH with IgH break-apart probe positive for IgH rearrangement. (C) Interphase FISH with CCND1::IgH dual color dual fusion probe positive for t(11;14). (D) Interphase FISH with LSI D5S23, D5S721/CEP 9/CEP 15 tricolor probe depicting trisomies for chromosomes 5,9,15. (E) Interphase FISH with 1p(CDKN2C)/1q(CKS1B) LSI probe depicting deletion of 1p(CDKN2C) and gain of 1q(CKS1B). (F) Interphase FISH targetingTP53 gene depicting TP53 deletion.
| Figure 3: Magnetic cell sorting (MACS) of CD138+ plasma cells using anti-CD138 and anti-dextran complex through positive selection of plasma cells in a magnetic chamber.
References
- 1 Campo E, Jaffe ES, Cook JR. et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 2022; 140 (11) 1229-1253
- 2 Swerdlow SH, Campo E, Pileri SA. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127 (20) 2375-2390
- 3 Kumar SK, Rajkumar V, Kyle RA. et al. Multiple myeloma. Nat Rev Dis Primers 2017; 3 (01) 17046
- 4 Rajan AM, Rajkumar SV. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J 2015; 5 (10) e365-e365
- 5 Furukawa Y, Kikuchi J. Molecular pathogenesis of multiple myeloma. Int J Clin Oncol 2015; 20 (03) 413-422
- 6 Cardona-Benavides IJ, de Ramón C, Gutiérrez NC. Genetic abnormalities in multiple myeloma: prognostic and therapeutic implications. Cells 2021; 10 (02) 336
- 7 Hanamura I. Multiple myeloma with high-risk cytogenetics and its treatment approach. Int J Hematol 2022; 115 (06) 762-777
- 8 Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol 2020; 95 (05) 548-567
- 9 D'Agostino M, Cairns DA, Lahuerta JJ. et al. Second revision of the International Staging System (R2-ISS) for overall survival in multiple myeloma: a European Myeloma Network (EMN) Report within the HARMONY Project. J Clin Oncol 2022; 40 (29) 3406-3418
- 9 Rajkumar SV, Tefferi A. Expanding the journal's circulation to oncologists—a new dimension for Mayo Clinic proceedings. Mayo Clin Proc 2007; 82 (08) 915-916
- 11 Russell SJ, Rajkumar SV. Multiple myeloma and the road to personalised medicine. Lancet Oncol 2011; 12 (07) 617-619
- 12 Rajkumar SV, Kohli M. Introduction to the symposium on neoplastic hematology and medical oncology. Mayo Clin Proc 2015; 90 (07) 848-849
- 13 Treatment-of-Newly-Diagnosed-Myelomav16_Nov19FINAL.pdf. Accessed October 10, 2022. Accessed Feb 1, 2023 at: https://static1.squarespace.com/static/5b44f08ac258b493a25098a3/t/5dd6c13ee9a64d38904eabda/1574355263029/Treatment-of-Newly-Diagnosed-Myelomav16_Nov19FINAL.pdf
- 14 Baysal M, Demirci U, Umit E. et al. Concepts of double hit and triple hit disease in multiple myeloma, entity and prognostic significance. Sci Rep 2020; 10 (01) 5991
- 15 Shen M, Yang G, Li X, Geng C, Huang Z, Chen W. At least two high-risk cytogenetic abnormalities indicate the inferior outcomes for newly diagnosed multiple myeloma patients: a real-world study in China. Leuk Lymphoma 2021; 62 (12) 2992-3001
- 16 Walker BA, Mavrommatis K, Wardell CP. et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019; 33 (01) 159-170
- 17 Talley PJ, Chantry AD, Buckle CH. Genetics in myeloma: genetic technologies and their application to screening approaches in myeloma. Br Med Bull 2015; 113 (01) 15-30
- 18 Stevens-Kroef M, Weghuis DO, Croockewit S. et al. High detection rate of clinically relevant genomic abnormalities in plasma cells enriched from patients with multiple myeloma. Genes Chromosomes Cancer 2012; 51 (11) 997-1006
- 19 Chng WJ, Dispenzieri A, Chim CS. et al; International Myeloma Working Group. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014; 28 (02) 269-277
- 20 Anguiano A, Tuchman SA, Acharya C. et al. Gene expression profiles of tumor biology provide a novel approach to prognosis and may guide the selection of therapeutic targets in multiple myeloma. J Clin Oncol 2009; 27 (25) 4197-4203
- 21 Abós B, Wang T, Castro R. et al. Distinct differentiation programs triggered by IL-6 and LPS in teleost IgM(+) B cells in the absence of germinal centers. Sci Rep 2016; 6 (01) 30004
- 22 Kishimoto RK, de Freitas SLVV, Ratis CA, Borri D, Sitnik R, Velloso EDRP. Validation of interphase fluorescence in situ hybridization (iFISH) for multiple myeloma using CD138 positive cells. Rev Bras Hematol Hemoter 2016; 38 (02) 113-120
- 23 López de Frutos L, Álvarez Y, Armengol G, Caballín MR. New mitogens in cultures for multiple myeloma cytogenetic analysis. Leuk Lymphoma 2013; 54 (11) 2548-2550
- 24 Sonneveld P, Avet-Loiseau H, Lonial S. et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood 2016; 127 (24) 2955-2962
- 25 Fonseca R, Bergsagel PL, Drach J. et al; International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia 2009; 23 (12) 2210-2221
- 26 Fonseca R, Barlogie B, Bataille R. et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res 2004; 64 (04) 1546-1558
- 27 Wahed A, Quesada A, Dasgupta A. . Application of flow cytometry in diagnosis of hematological disorders. In: Hematology and Coagulation. Elsevier; 2020:127–141.
- 28 Hartmann L, Biggerstaff JS, Chapman DB. et al. Detection of genomic abnormalities in multiple myeloma: the application of FISH analysis in combination with various plasma cell enrichment techniques. Am J Clin Pathol 2011; 136 (05) 712-720
- 29 Khoenkhoen S, Ádori M, Pedersen GK, Karlsson Hedestam GB. Flow cytometry-based protocols for the analysis of human plasma cell differentiation. Front Immunol 2020; 11: 571321
- 30 Mercer BR, Rayeroux KC. . Detection of chromosome abnormalities using cytoplasmic immunoglobulin staining and FISH in myeloma. In: Campbell LJ, ed. Cancer Cytogenetics. Vol. 730. Methods in Molecular Biology. Humana Press; 2011:159–171. doi:
- 31 Yang HS, Montella J, Holt L. et al. Cytoplasmic immunoglobulin fluorescence in situ hybridization (cIg FISH) enhances the quantitative detection of chromosome abnormalities in plasma cell neoplasms compared to conventional FISH. Blood 2010; 116 (21) 1198-1198
- 32 Gole L, Lin A, Chua C, Chng WJ. Modified cIg-FISH protocol for multiple myeloma in routine cytogenetic laboratory practice. Cancer Genet 2014; 207 (1-2): 31-34
- 33 Ma ESK, Wang CLN, Wong ATC, Choy G, Chan TL. Target fluorescence in-situ hybridization (target FISH) for plasma cell enrichment in myeloma. Mol Cytogenet 2016; 9 (01) 63
- 34 Yan Y, Qin X, Liu J. et al. Clonal phylogeny and evolution of critical cytogenetic aberrations in multiple myeloma at single-cell level by QM-FISH. Blood Adv 2022; 6 (02) 441-451
- 35 Ross FM, Avet-Loiseau H, Ameye G. et al; European Myeloma Network. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica 2012; 97 (08) 1272-1277
- 36 Ciolino AL, Tang ME, Bryant R. Statistical treatment of fluorescence in situ hybridization validation data to generate normal reference ranges using Excel functions. J Mol Diagn 2009; 11 (04) 330-333
- 37 Kadam Amare PS, Jain H, Nikalje S. et al. Observation on frequency & clinico-pathological significance of various cytogenetic risk groups in multiple myeloma: an experience from India. Indian J Med Res 2016; 144 (04) 536-543
- 38 Nishida K, Tamura A, Nakazawa N. et al. The Ig heavy chain gene is frequently involved in chromosomal translocations in multiple myeloma and plasma cell leukemia as detected by in situ hybridization. Blood 1997; 90 (02) 526-534
- 39 Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front Immunol 2019; 10: 1121
- 40 Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 2017; 14 (02) 100-113
- 41 Kumar S, Kaufman JL, Gasparetto C. et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017; 130 (22) 2401-2409
- 42 Pawlyn C, Davies FE. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019; 133 (07) 660-675
- 43 Cazaubiel T, Leleu X, Perrot A. et al. Primary plasma cell leukemias displaying t(11;14) have specific genomic, transcriptional, and clinical features. Blood 2022; 139 (17) 2666-2672
- 44 Kalff A, Khong T, Wall M. et al. A rare case of IGH/MYC and IGH/BCL2 double hit primary plasma cell leukemia. Haematologica 2015; 100 (02) e60-e62
- 45 Kumar S, Fonseca R, Ketterling RP. et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2012; 119 (09) 2100-2105
- 46 Chretien ML, Corre J, Lauwers-Cances V. et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter?. Blood 2015; 126 (25) 2713-2719
- 47 Caers J, Garderet L, Kortüm KM. et al. European Myeloma Network recommendations on tools for the diagnosis and monitoring of multiple myeloma: what to use and when. Haematologica 2018; 103 (11) 1772-1784
- 48 Ye JC, Chen L, Chen J. et al. Aneuploidy is associated with inferior survival in relapsed refractory multiple myeloma patients. Blood 2019; 134 (Supplement_1): 4360-4360
- 49 Schmidt TM, Barwick BG, Joseph N. et al. Gain of chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J 2019; 9 (12) 94
- 50 Bisht K, Walker B, Kumar SK. et al. Chromosomal 1q21 abnormalities in multiple myeloma: a review of translational, clinical research, and therapeutic strategies. Expert Rev Hematol 2021; 14 (12) 1099-1114
- 51 D'Agostino M, Ruggeri M, Aquino S. et al. Impact of gain and amplification of 1q in newly diagnosed multiple myeloma patients receiving carfilzomib-based treatment in the forte trial. Blood 2020; 136 (Supplement (Suppl. 01) 38-40
- 52 Treon SP, Maimonis P, Bua D. et al. Elevated soluble MUC1 levels and decreased anti-MUC1 antibody levels in patients with multiple myeloma. Blood 2000; 96 (09) 3147-3153
- 53 Sawyer JR, Tricot G, Lukacs JL. et al. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer 2005; 42 (01) 95-106
- 54 Legartova S, Krejci J, Harnicarova A, Hajek R, Kozubek S, Bartova E. Nuclear topography of the 1q21 genomic region and Mcl-1 protein levels associated with pathophysiology of multiple myeloma. Neoplasma 2009; 56 (05) 404-413
- 55 Hebraud B, Leleu X, Lauwers-Cances V. et al. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: the IFM experience on 1195 patients. Leukemia 2014; 28 (03) 675-679
- 56 Avet-Loiseau H, Li C, Magrangeas F. et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol 2009; 27 (27) 4585-4590
- 57 Boyd KD, Ross FM, Walker BA. et al; NCRI Haematology Oncology Studies Group. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clin Cancer Res 2011; 17 (24) 7776-7784
- 58 Corre J, Perrot A, Caillot D. et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood 2021; 137 (09) 1192-1195
- 59 Merz M, Hielscher T, Seckinger A. et al. Baseline characteristics, chromosomal alterations, and treatment affecting prognosis of deletion 17p in newly diagnosed myeloma. Am J Hematol 2016; 91 (11) E473-E477
- 60 Fonseca R, Oken MM, Harrington D. et al. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia 2001; 15 (06) 981-986
- 61 Weinhold N, Kirn D, Seckinger A. et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica 2016; 101 (03) e116-e119
- 62 Glitza IC, Lu G, Shah R. et al. Chromosome 8q24.1/c-MYC abnormality: a marker for high-risk myeloma. Leuk Lymphoma 2015; 56 (03) 602-607
- 63 Abdallah N, Baughn LB, Rajkumar SV. et al. Implications of MYC rearrangements in newly diagnosed multiple myeloma. Clin Cancer Res 2020; 26 (24) 6581-6588
- 64 Caracciolo D, Scionti F, Juli G. et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2021; 106 (01) 185-195
- 65 Kim H, Moon HW, Hur M, Yun YM, Park CM, Lee MH. Variant Burkitt-type translocation (8;22)(q24;q11) in plasma cell myeloma. Korean J Hematol 2011; 46 (02) 135-138
- 66 Kyle RA, Therneau TM, Rajkumar SV. et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346 (08) 564-569
- 67 Lakshman A, Paul S, Rajkumar SV. et al. Prognostic significance of interphase FISH in monoclonal gammopathy of undetermined significance. Leukemia 2018; 32 (08) 1811-1815
- 68 Kyle RA, Remstein ED, Therneau TM. et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med 2007; 356 (25) 2582-2590
- 69 Mateos MV, Kumar S, Dimopoulos MA. et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J 2020; 10 (10) 102
- 70 Rajkumar SV, Gupta V, Fonseca R. et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013; 27 (08) 1738-1744
- 71 Neben K, Jauch A, Hielscher T. et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol 2013; 31 (34) 4325-4332
- 72 Papadhimitriou SI, Terpos E, Liapis K. et al. The cytogenetic profile of primary and secondary plasma cell leukemia: etiopathogenetic perspectives, prognostic impact and clinical relevance to newly diagnosed multiple myeloma with differential circulating clonal plasma cells. Biomedicines 2022; 10 (02) 209
- 73 Mina R, D'Agostino M, Cerrato C, Gay F, Palumbo A. Plasma cell leukemia: update on biology and therapy. Leuk Lymphoma 2017; 58 (07) 1538-1547
- 74 Fernández de Larrea C, Kyle R, Rosiñol L. et al. Primary plasma cell leukemia: consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J 2021; 11 (12) 192
- 75 Janssen JW, Vaandrager JW, Heuser T. et al. Concurrent activation of a novel putative transforming gene, myeov, and cyclin D1 in a subset of multiple myeloma cell lines with t(11;14)(q13;q32). Blood 2000; 95 (08) 2691-2698