 PDF
PDF  Views
Views  Share
Share


Rare childhood tumors in a Turkish pediatric oncology center
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2013; 34(04): 264-269
DOI: DOI: 10.4103/0971-5851.125241
Abstract
Background: It has been estimated that rare tumor rate is about 15% of all childhood cancer in United States. According to Turkish Pediatric Oncology Group (TPOG) datas, 8889 children were diagnosed between 2002 and 2008 in our country and 3.7% of them were diagnosed as rare tumors. Aim: To investigate the frequency and clinical features of rare tumors in our pediatric oncology center. Materials and Methods: A total of 43 cases that have diagnosed as rare tumor in 574 cancer patients between the yaer 2002 and 2012 were reviewed retrospectively. All cases definitive diagnosis were established by histopathological and immunohistochemical studies. Results: Frequency of rare tumors was 7.4% in our center. Benign and border line rare tumors were 27 (62.7%) cases, malignant rare tumor were 16 (37.2%) cases. Median follow-up period was 48 months (between 1 and 110 months). Six of the malignant rare tumors were died with progressive disease (synovial sarcoma, mixed malignant mesenchymal tumor, undifferentiated sarcoma, plexus choroideus carcinoma, renal peripheral primitive neuroectodermal tumor, adrenocortical carcinoma). Malignant rare tumor mortality rate was found 37.5% in our clinic. Conclusion: We have found that our rare tumor rate (7.4%) was higher than Turkish rare tumor rate (3.7%) according to TPOG′s datas. However, it was still lower than rare tumor rates of western countries (15%), probably due to difficulties of diagnosis and referral problems.
Publication History
Article published online:
19 July 2021
© 2013. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Background:
It has been estimated that rare tumor rate is about 15% of all childhood cancer in United States. According to Turkish Pediatric Oncology Group (TPOG) datas, 8889 children were diagnosed between 2002 and 2008 in our country and 3.7% of them were diagnosed as rare tumors.
Aim:
To investigate the frequency and clinical features of rare tumors in our pediatric oncology center.
Materials and Methods:
A total of 43 cases that have diagnosed as rare tumor in 574 cancer patients between the yaer 2002 and 2012 were reviewed retrospectively. All cases definitive diagnosis were established by histopathological and immunohistochemical studies.
Results:
Frequency of rare tumors was 7.4% in our center. Benign and border line rare tumors were 27 (62.7%) cases, malignant rare tumor were 16 (37.2%) cases. Median follow-up period was 48 months (between 1 and 110 months). Six of the malignant rare tumors were died with progressive disease (synovial sarcoma, mixed malignant mesenchymal tumor, undifferentiated sarcoma, plexus choroideus carcinoma, renal peripheral primitive neuroectodermal tumor, adrenocortical carcinoma). Malignant rare tumor mortality rate was found 37.5% in our clinic.
Conclusion:
We have found that our rare tumor rate (7.4%) was higher than Turkish rare tumor rate (3.7%) according to TPOG's datas. However, it was still lower than rare tumor rates of western countries (15%), probably due to difficulties of diagnosis and referral problems.
INTRODUCTION
Unlike adults, It is well known that childhood cancer is seen rarely in the world, with annual incidence usually between 70/million to 160/million at the age of 0-14 years.[1] In Turkey, the frequency of cancer is between 110/million and 150/million in children under the age of 15 years.[2,3] In childhood and adolescence, a wide variety of rare tumors can occur with particular biological and clinical characteristics, although the great variations are seen between populations for some specific tumour types.[4] These tumors can be rare in any age group common tumors of adults can appear in childhood and adolescence and many rare tumors and tumor-like lesions show a predilection for younger age groups. Also, frequent tumors can be present with rare histologic features or can occur in rare atypical locations.[5] According to TREP Project in Italy; Rare tumors were defined as that those malignancies characterized by an annual incidence of < 2/million children and adolescents up to 18 years old of age.[4,5,6,7]
To diagnose and manage these rare tumors could be difficult for developing countries due to technical difficulties. In this study, we aimed to analyze the frequencies, types and clinical features of our rare tumors in our center.
MATERIALS AND METHODS
Files of the all patients who were diagnosed as a childhood cancer have been analyzed retrospectively. 574 patients that have been diagnosed as childhood cancers between 2002 and 2012 years have enrolled in this study. 43 of 574 patients had been diagnosed as rare tumors. The definitive diagnosis of patients were established by histopathological and immunohistochemical investigations. Patients age, gender, stages, histopathological types of tumor, clinical features and mortality rates were recorded.
Statistical analysis
Mortality rates were analyzed with the Kaplan–Meier method and other analysis were estimated using descriptive statistics with SPSS 15.0 (SPSS Inc, Chicago, IL, USA) statistics program for windows.
RESULTS
Fourty three of 574 patients had been diagnosed as rare tumors in 10 years period in our clinic. Their ages at the time of initial diagnosis were between 1 month and 18 years old (median: 10 years). Around 22 of them were boys and 21 were girls. Benign and border line rare tumors were 27 (62.7%) cases, malignant rare tumors were 16 (37.2%) cases. Rare tumors consisted of papillary thyroid carcinoma, medullary thyroid carcinoma, giant cell granuloma, carcinoid tumor, thymic carcinoma, synovial sarcoma, nasopharyngeal carcinoma, optic glioma, myxofibrosarcoma, primary bone rhabdomyosarcoma, chest wall primitive neuroectodermal tumor (PNET), renal PNET, mixed malignant mesenchymal tumor, carcinoma of choroid plexus, undifferentiated sarcoma, hepatoblastoma, adrenocortical carcinoma, primary gastric carcinoma, intrarenal pheochromocytoma, sertoli cell tumor, cystic lymphangioma, giant congenital nevus, atypical fibroxanthoma, juvenile xanthogranuloma, desmoid tumor, neurofibroma, cystic hygroma, orbital lymphangioma, multiple hereditary exostoses syndrome, Type I neurofibromatosis (NFI) and schwannoma [Tables [Tables11 and and22].
Table 1
Types of our bening and border line rare tumors
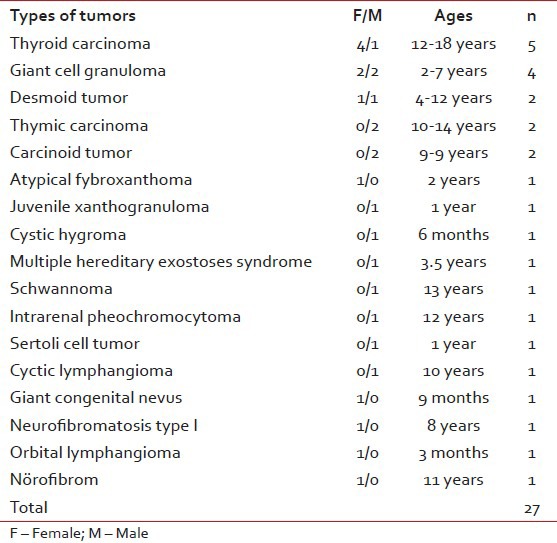
Table 2
Tumor types of our malignant rare tumors
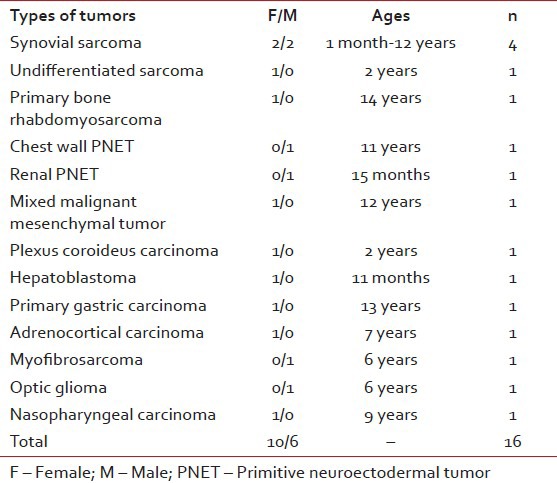
Frequency of rare tumors was 7.4% in our center. About16 (37.2%) of rare tumors were malignant, the other were benign or borderline cases. Median follow-up period was 48 months (ranges 1-110 months).
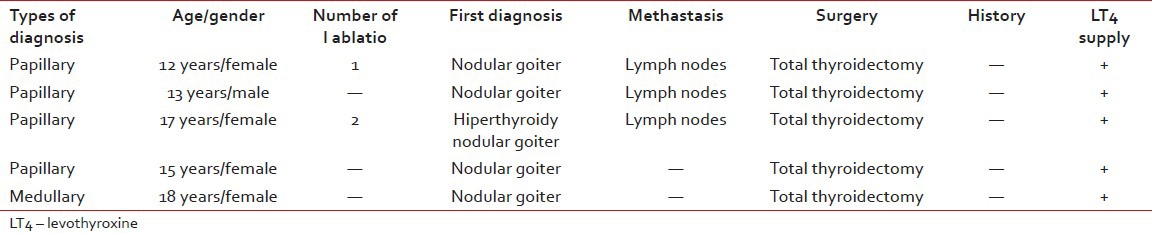
Thyroid carcinomas were the most common tumor types of all rare tumors (11.6%). Four of them were papillary thyroid carcinoma (%80). The other was medullary thyroid carcinoma. Their ages were between 12 and 18 years old age (median: 15 years). Four patients were female (%80), one patient was male. Three of all cases have methastasis at the time of initial diagnosis (%60). All of them had treated with levothyroxine after total thyroidectomy [Table 3].
Table 3
Clinical features of thyroid cancer patients
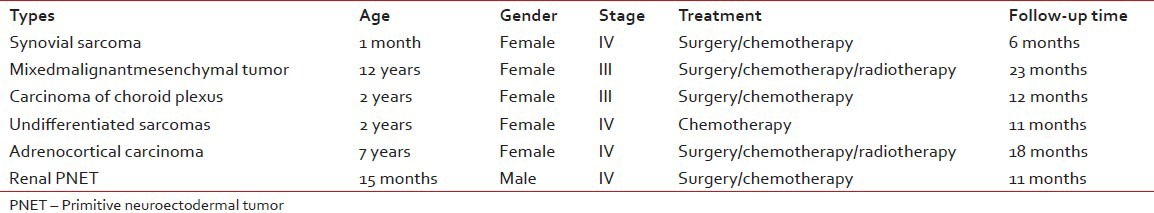
Six of the malignant rare tumors (37.5%) died with progressive disease. These cases were synovial sarcoma, mixed malignant mesenchymal tumor, undifferentiated sarcoma, plexus coroideus carcinoma, adrenocortical carcinoma and renal PNET [Table 4]. Their ages were between 1 month and 12 years old ages (median: 4, 5 years). One of them was male and the others were female. Follow-up time period was between 6 and 23 months (median: 11.5 months). All cases were being in advanced stages at the time of initial diagnosis.
Table 4
Clinical features of the patients who died with progressive disease
DISCUSSION
It is well known, childhood cancer is relatively uncommon comparing with adults. Because of its rarity, epidemiologic studies of childhood cancer are also challenging. According to Rare Tumors Committee datas, It has been estimated that rare tumour rate is about 15% of all childhood cancer in United States.[1,4,5,6,7,8,9,10] According to Turkish Pediatric Oncology Group (TPOG) datas, 8889 children between 0 and 18 years of ages diagnosed as childhood cancers between 2002 and 2008 in our country and the rare tumour rate was 3.7%.[3] We have determined the frequency of rare tumors 7.4% in our center between 2002 and 2012. This rate were higher than TPOG's datas. It was concluded that most of patient were referred from other hospitals, because of technical difficulties of diagnosis and failure of experienced health team. However, our rare tumors rate is still lower than western countries.
In 2000, the TREP (rare tumours in pediatric age) project, a nation-wide Italian cooperative project, was launched in Italy to improve basic research on these tumors and their clinical management. The project aimed to establish a network of pediatric and adult oncologists, surgeons, pathologists and biologists, to pool clinical data and develop diagnostic and therapeutic recommendations for each of these rare tumors. Nasopharyngeal carcinoma, adrenocortical tumors, pleuropulmonary blastoma and other lung tumors, carcinoid tumors, cutaneous melanoma, renal carcinoma, pancreatoblastoma and other pancreatic exocrine tumors, gonadal non-germ-cell tumors (ovary/testis), pheochromocytoma, paraganglioma, thyroid carcinoma, salivary gland tumors, breast carcinoma, gastrointestinal tract carcinoma and thymus carcinoma were enrolled into this study group.[6,7] Our rare tumors list were different from TREP list as we have included the cases that we had seen rarely, like hepatoblastom, renal PNET, adrenocortical carcinoma, synovial sarcoma etc.
Thyroid carcinoma, carcinoma of appendix and gonadal non-germ-cell tumors were the larger groups in the TREP project, 19.2%, 18.8%, 10.3% respectively.[6,7] In our study, the most common tumor types among adolescents in our rare tumor cases were also thyroid carcinomas (11.6%) like TREP list. Although incidence seldom exceeds 1.5/million, the most common carcinoma in children is the thyroid carcinoma in many world regions. An enormous increase in thyroid carcinoma was observed in areas of Belarus, Ukraine and Russia most heavily contaminated by fallout from Chernobyl.[11] The United Kingdom Children's Cancer Study Group and the US Children's Oncology Guidelines recommend yearly thyroid palpation followed by ultrasound or fine needle aspiration if there is an abnormality.[12]
The central giant cell granuloma (CGCG) was the second largest group with four cases in our series (9.3%). Two cases of CGCG were siblings. CGCG is a benign lesion of the jaw of unknown etiology. It is found predominantly in children and young adults. Lesions are more common in the anterior region of the jaws and mandibular lesions frequently extend across the midline.[13]
In our study, synovial sarcoma cases were the other second larger group (9.3%). Synovial sarcoma is thought to arise from primitive mesenchymal cells rather than from the synovial membrane as its name implies. Although these tumors can occur anywhere in the body, including locations distant from joint spaces such as the abdominal cavity, approximately 90% of synovial sarcomas in children occur in the extremities.[14]
Our cases of carcinoid tumors had been diagnosed after appendectomy. These cases were the third larger group (4.6%) in our rare tumor list where as the second larger group in TREP list (18.8%). Carcinoid tumor of the appendix has been known since 1808 and currently we know that it may appear all along the intestinal tract, biliary tree, ovaries, bronchi, lungs and pancreas. It is usually uncommon during infancy, representing 1.8% of all the tumors of the intestinal tract, approximately 70% of the appendiceal tumors and 0.16-0.7% of all operated appendixes. When the diameter is < 1 cm, the treatment of choice is appendectomy; and in tumors of larger size, surgery is more extensive.[15]
Another third larger group was thymic tumour group (4.6%) in our list. Thymic tumours are exceptionally rare in the pediatric age group, accounting for < 1% of childhood mediastinal tumours. As in adults, thymic tumours in children may be asymptomatic or present with compressive symptoms.[16] Like timic and carcinoid tumor, our desmoid tumor frequency was found 4.6% of our tumor list. Desmoid tumors are uncommon fibroblastic proliferative lesions and the primary treatment of these lesions is surgical resection. Careful postoperative surveillance is warranted, because these tumors have a high rate of recurrence.[17]
The nasopharyngeal carcinoma[18] is very rare in childhood and differs from its adult counterpart in the prevalence of the non-keratinizing, undifferentiated subtype and by an advanced clinical stage at onset and better chances of survival; peripheral PNET of the chest wall[19] is a rare malignant tumor that shares a similar histology, immunohistology and cytogenetics to Ewing's sarcoma and usually occurring in children; renal PNET[20] are also exceptional and belong to the vast group of peripheral malignant primary neuroectodermal tumours; mixed malignant mesenchymal tumors[21,22,23] are also rare sarcoma that have been reported that originating from the cerebellar vermis, uterine tube and breast; undifferentiated sarcomas[24] are primitive mesenchymal tumors that can not be classified among standardized histopathologic entities; primary bone rhabdomyosarcoma[25] is also rarely seen in childhood; although hepatoblastoma[26] is the most common primary hepatic malignancy in childhood, it is quite rare and accounting for only 0.9% of all pediatric cancers; choroid plexus carcinomas[27] are rare tumors with dismal prognosis; primary gastric carcinoma[28] accounts for only 0.05% of pediatric gastrointestinal malignancies and its, presentation are similar to those of adult gastric carcinoma, intrarenal pheochromocytoma[29] (paraganglioma) is a very rare tumour and its diagnosis is often difficult; sertoli cell tumors[30] are frequently benign rare tumors and are seen in children before 1 year of age; cystic lymphangioma[31] is an uncommon congenital neoplasm which frequently occurs in young adults and < 1% are localized in the mediastinum; giant congenital nevi[32] are seen rarely and disfiguring, potentially malignant pigmented nevi present at birth; atypical fibroxanthoma[33] is a nodular dermal ulcerative lesion with a favorable prognosis and most commonly occurs on sun-exposed skin in elderly individuals, neurofibromatosis (NFI)[34] is an autosomal dominant disorder with a prevalence about 1/3000 (1/2000-1/5000 in various population-based studies) and about 15% children with NFI develop optic pathway glioma[35] so the neurofibromatosis society recommend annual ophthalmic screening; juvenile xanthogranuloma[36] is one of the most common forms of non-Langerhans cell histiocytosis in children, although it usually presents as a self-limited skin lesion with typical histopathology, it can be challenging to diagnose due to an atypical initial presentation; orbital lymphangioma[37] is an uncommon, benign cystic lesion and usually presents with a slowly progressive proptosis, displacement of the globe, ptosis and restriction of eye movements in childhood; schwannomas[38] are being the most frequently encountered neurogenic tumours type of the posterior mediastinum that can be either benign or malignant, although the former is more common; hereditary multiple exostoses[39] is an autosomal dominant condition characterized by growth of multiple benign cartilage-capped tumors and greatly increases the relative risk to develop chondrosarcoma although most chondrosarcomas are sporadic; adrenocortical carcinoma[40] is a rare tumor and its incidence is < 0.5/million among children however much more common in southern Brazil with an incidence of at least 1.5/million in Sao Paulo and Parana; neurofibroma[41] is a benign tumor of the peripheral nerve sheath characterized by proliferation of Schwann's cells, perineural cells and endoneurial fibroblasts; myofibrosarcoma[42] is a distinctive malignant soft tissue neoplasm and its histology ranges between low-grade spindle cell to high-grade fibrohistiocytic histologies and often has myoid characteristics.
As a result of improved survival, the cancer mortality rates have decreased for children since the 1950's and by the late 1990's had decreased to < 30/million.[9] Rare tumors, when considered as a group, represent a significant burden to society as they may account for up to 25% of the mortality by cancer in nations like the United States. In contrast with the current scenario in highly incident cancer types, little progress has been achieved in the treatment of the most rare cancers. The reasons for this apparent stagnation are mostly intrinsic to logistical difficulties in performing large clinical trials in rare diseases.[43] Our mortality rate of all pediatric cancer patients in our clinic was estimated to 33.8% between 2002 and 2008 years according to TPOG's datas. The mortality rate of our malignant rare tumors (37.5%) between 2002 and 2012 was more higher than both our mortality rates of all childhood tumors and developed country rare tumor mortality rates. To our knowledge so far the studies about rare tumors had not been conducted in developing countries so it couldn’t be possible to compare our results with developing countries datas. It was concluded that in spite of multi-modal therapies and technical development for diagnostic intervention, management of rare tumors is difficult for developing countries.
CONCLUSION
It has been found that the rate of our rare tumors are higher than TPOG's datas but lower than developed countries rate. As our rare tumor list were comprise of benign, borderline and malignant neoplasms like TREP list, benign or borderline cases may have been giving rise to the high ratio observed in our clinic comparing with TPOG's datas. Also, the reason of the rare tumors lower rate than developed countries rate may be related to unable to acces to pediatric oncologist and hospital care, due to economical and techinical difficulties so their diagnosis, follow-up and treatment is still a big health problem in developing countries.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.