 PDF
PDF  Views
Views  Share
Share


Primary extraosseous intracranial Ewing?s sarcoma: Case report and literature review
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(02): 118-121
DOI: DOI: 10.4103/0971-5851.89798
Abstract
Ewing′s sarcoma / peripheral primitive neuroectodermal tumors occur most often in bone and soft tissues of children and young adults. The intracranial manifestation of the disease is rare, and when present, this is often misdiagnosed with other varieties of primary brain tumors. We report such a case of extraosseous Ewing′s sarcoma, which was initially suspected to be a case of meningioma in an 11-year-old girl.
Publication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Ewing's sarcoma / peripheral primitive neuroectodermal tumors occur most often in bone and soft tissues of children and young adults. The intracranial manifestation of the disease is rare, and when present, this is often misdiagnosed with other varieties of primary brain tumors. We report such a case of extraosseous Ewing's sarcoma, which was initially suspected to be a case of meningioma in an 11-year-old girl.
INTRODUCTION
Ewing's sarcoma family of tumors are a group of small round-cell neoplasms, which include Ewing's sarcoma (EWS), Primitive neuroectodermal tumor (PNET), Askins tumor, PNET of the bone, and Extraosseous Ewing's sarcoma (ESS).[1] Ewing's sarcoma / peripheral primitive neuroectodermal tumor, presumed to be neuroectodermal in origin, most often occurs in the bone and soft tissues of children and young adults. The intracranial Extraosseous Ewing's sarcoma (CNS-ESS) is extremely rare and often misdiagnosed as central nervous system PNET (c-PNET) or as other primary intracranial neoplasms.
CASE REPORT
An 11-year-old female presented with a history of headache for one year, which increased in intensity over the last one month. She had repeated episodes of vomiting for one month and a left temporal scalp swelling, which was gradually increasing in size. There was no history of loss of consciousness, limb weakness or seizure. On examination there was bilateral papilledema, but no other focal neurological deficits were detected. The left temporal scalp swelling was mildly tender, firm, immobile, and non-pulsatile with ill-defined margins.
The Computed Tomography Scan (CT Scan) of the brain showed a rounded, well-defined, heterogeneously hyperdense, enhancing lesion in the left temporoparietal region, with a mass effect and destruction of the left temporal bone, extending into the scalp, suggesting the possibility of a meningioma. No evidence of calcification was noted within the lesion [Figure 1]. Magnetic resonance imaging (MRI) of the brain showed a left temporal lesion, hypointense on T1, heterointense on T2, with heterogeneous enhancement [Figure 2]. The lesion measured 6.0×4.6×7.4 cm. A significant mass effect was detected with a midline shift to the right, of 15 mm. The diffusion weighted image showed mild restriction. Magnetic resonance spectroscopy showed choline and N-Acetyl Acetate peaks.
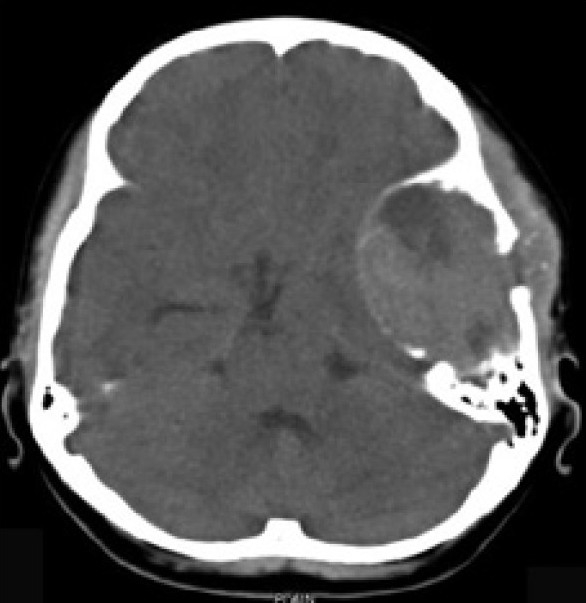
| Fig. 1 Preoperative CT scan of the brain showed a rounded, well-defined, heterogeneously hyperdense, enhancing lesion in the left temporoparietal region, with a mass effect and destruction of the left temporal bone extending into the scalp, suggesting the possibility of meningioma. No evidence of calcification was noted within the lesion
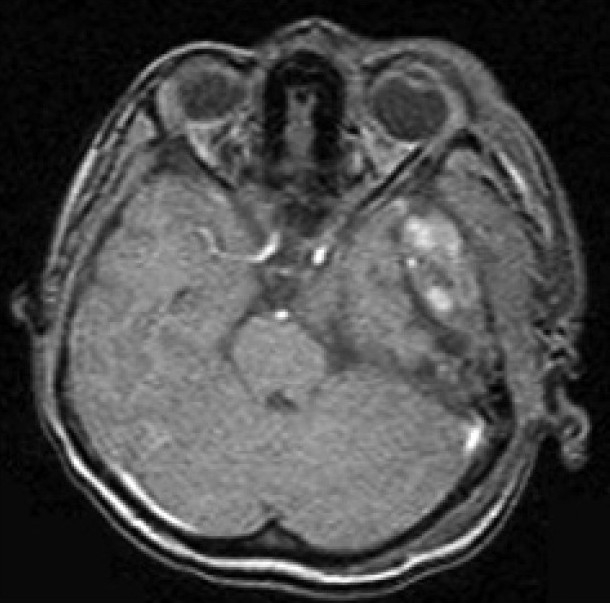
| Fig. 2 Preoperative MRI of the brain showed a left temporal lesion, hypointense on T1, heterointense on T2, with heterogenous enhancement
The patient underwent left temporoparietal craniotomy and decompression of the lesion. The operative findings suggested that the lesion was extradural, with erosion of bone in some areas and involvement of the overlying temporalis muscle.
The histopathological examination showed round-to-oval cells arranged in lobules, separated by a thin vascular channel, having vesicular nuclei, with indistinct nucleoli. Mitotic figures were seen with focal areas of necrosis. A few bony trabeculae were seen embedded in the tumor island. Fat globules seen were positive for Periodic Acid Schiff's staining. Immunohistochemistry showed focal positivity for synaptophysin and MIC-2 [Figure 3]. It was negative for CD20, CD3, Myosin, and Glial Fibrillary Acidic Protein. The overall features were compatible with a Primitive Neuroectodermal Tumor (PNET), with a possibility of Ewing's sarcoma in the temporal region . The tumor was positive for t (11; 22) (q24; q12) translocation detected by fluorescent in situ hybridization (FISH) in the tumor biopsy sample. The findings of further metastatic workups, including CT Scans of the thorax and abdomen and a bone scan with Technetium Tc-99m, were negative.
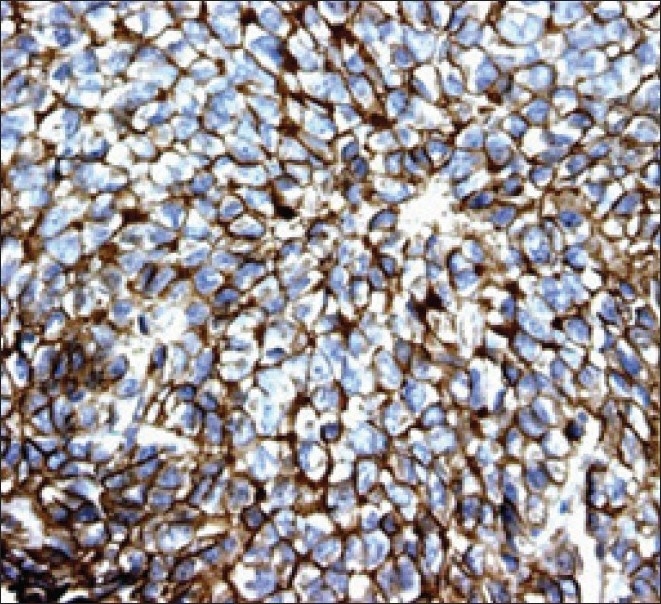
| Fig. 3 EWS / PNET's are characterized by immunoreactivity to the surface antigen CD99 / MIC2, which is expressed in up to 97% of the cases
A postoperative CT scan of the brain revealed a large extradural and subdural hematoma in the left frontotemporal region, with a postoperative craniotomy defect and scalp edema. The child went on to receive 12 weeks of chemotherapy with vincristine, doxorubicin, and cyclophosphamide, alternated with ifosfamide and etoposide. Radiation with a dose of 56 Gray was prescribed at the local site. The treatment was completed with the same combination of chemotherapeutic drugs for another 24 weeks. The child is doing well and is under post treatment surveillance.
DISCUSSION
James Ewing (1866–1943) first described the tumor, establishing that the disease was different from lymphoma and other types of cancer known at that time.[2] In 1921, he described a lethal primary bone lesion that affects children and young adults and most frequently originates in the long bones (47%), pelvis (19%) or ribs (12%).[3] The skull is rarely involved, probably in less than 4% of the cases, with the frontal and parietal bones being the most commonly affected.[4,5]
Extraosseous Ewing's sarcoma (EES) has been recognized as a distinct disease entity that afflicts young adults in the second and third decades of life, with equal sex predilection. The EES commonly involves the paravertebral regions of the spine and in rare instances, these lesions arise in the intracranial compartment, where they have been commonly misdiagnosed as c-PNET, because of the similarity in their histological appearance.[6]
Very few cases of the central nervous system extraosseous Ewing's sarcoma (CNS-EES) have been reported in pathology literature.[6–11] Jay et al. was probably the first to describe a patient with an isolated posterior fossa mass that histologically resembled a medulloblastoma, but demonstrated the t(11;22) (q24;q12) translocation, which confirmed CNS-EES.[7] As far as our knowledge goes, this is the seventh case we are reporting.
Diagnosis requires a histopathological examination, immunohistochemistry, and cytogenetics. The differential diagnosis of an intracranial round cell tumor is primitive neuroectodermal tumor (neuroblastoma), lymphoma, rhabdomyosarcoma, and Ewing's sarcoma.
The histological examination reveals that these tumors are composed of small, undifferentiated neuroectodermal cells and frequently demonstrate immunohistochemical and/or electron microscopic features of glial or neuronal differentiation.[12] Recent advances in the molecular classification has allowed a clear pathological distinction between c-PNET and CNS-EES. CNS-EES is known to demonstrate in 97% of the patients, a strong membrane expression of the MIC-2 gene product, designated CD99, which is specifically recognized by the monoclonal antibodies O13 and HBA71.[13–16] In addition, the chromosomal translocation t(11, 22)(q24;q12), detected by FISH, is found in more than 90% of EES. This nonrandom translocation is not found in the central primitive neuroectodermal tumors (c-PNET) such as the medulloblastoma and supratentorial PNET.
Although, CNS-EES is histologically similar to c-PNETs such as the medulloblastoma, it differs significantly in clinical behavior, treatment, and prognosis.[17,18] The treatment options available for patients with CNS-EES are similar to those of EES elsewhere in the body and include multimodality treatment comprising of surgery, chemotherapy, and radiation. Patients with c-PNET also require surgery; however, the chemotherapeutic and radiation therapy protocols differ from those used for CNS-EES. Chemotherapy forms the backbone of the multimodality treatment of localized Ewing's sarcoma. Multiagent chemotherapy regimens including cyclophosphamide, ifosfamide, doxorubicin, dactinomycin, and etoposide have been shown to be effective in localized Ewing's sarcoma, in various trials.[19,20] The primary treatment for localized ESS consists of neoadjuvant chemotherapy, with a combination of vincristine, doxorubicin, and cyclophosphamide, alternating with ifosfamide and etoposide, given for 12 to 24 weeks, followed by definitive local treatment of surgery or radiation, or surgery and postoperative radiation directed at the primary site.[21] The treatment is completed with administering chemotherapy with a similar combination for a total of 36 to 49 weeks. Because of the small number of patients, the prognosis of CNS-EES is not clearly known, although it has been suggested that patients with EES that arises from structures within or around the CNS may have a more favorable outcome than patients with c-PNET.[22]
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Preoperative CT scan of the brain showed a rounded, well-defined, heterogeneously hyperdense, enhancing lesion in the left temporoparietal region, with a mass effect and destruction of the left temporal bone extending into the scalp, suggesting the possibility of meningioma. No evidence of calcification was noted within the lesion
| Fig. 2 Preoperative MRI of the brain showed a left temporal lesion, hypointense on T1, heterointense on T2, with heterogenous enhancement
| Fig. 3 EWS / PNET's are characterized by immunoreactivity to the surface antigen CD99 / MIC2, which is expressed in up to 97% of the cases