 PDF
PDF  Views
Views  Share
Share


Pheochromocytoma in a Child without Hypertension: A Contribution to the “Rule of 10s”
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(01): 59-61
DOI: DOI: 10.4103/0971-5851.203503
Abstract
Pheochromocytoma (PCC) is a neuroendocrine tumor originating from chromaffin tissue in adrenal medulla. Its diagnosis and treatment are well defined in adults, but experience in children is limited. Children constitute only 10% of reported cases, the average age at presentation being 11 years. The most common presentation is sustained hypertension, which is absent in only 10% of children. We managed a 14-month-old female child with PCC, but she was not hypertensive. We report two unusual features, in this case, an extremely young age at presentation and a childhood case of nonhypertensive PCC contributing for “rule of 10s.”
Publication History
Article published online:
06 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Pheochromocytoma (PCC) is a neuroendocrine tumor originating from chromaffin tissue in adrenal medulla. Its diagnosis and treatment are well defined in adults, but experience in children is limited. Children constitute only 10% of reported cases, the average age at presentation being 11 years. The most common presentation is sustained hypertension, which is absent in only 10% of children. We managed a 14-month-old female child with PCC, but she was not hypertensive. We report two unusual features, in this case, an extremely young age at presentation and a childhood case of nonhypertensive PCC contributing for “rule of 10s.”
Introduction
Pheochromocytoma (PCC) is a tumor of adrenal medulla chromaffin cells. When such tumors arise from extraadrenal chromaffin cells, they are called as paraganglioma (PGL). The reported incidence of PCC is 1/100,000 and 10% of cases occur in children.[1] The average age at diagnosis in children is 11 years.[2] Adrenal medulla is the major source of catecholamine which manifests as hypertension in patients with PCC.[3] In children, hypertension is detected in about 90% of cases and tends to be more severe and sustained than adults.[4,5] The purpose of reporting this case of PCC is because of its two unusual findings, namely very young age at presentation and childhood case without hypertension; each accounts for 10% of cases of PCC thus being categorized as “rule of 10s” or “10% tumor.”
Case Report
A 14-month-old female child presented to us for swelling of abdomen for last 1 month. Abdominal examination revealed a mass in the right hypochondrium and lumbar area which was nontender, a firm with an irregular surface. The blood pressure was normal. All the hematologic parameters were within normal limits. Ultrasonography suggested heterogeneous mixed echogenic solid lesions with the minimal cystic part within size 9 cm × 7 cm seen in the right hypochondrium and lumbar region. Computed tomography (CT) scan revealed 7.3 cm × 8.0 cm × 9.1 cm heterogeneous enhancing lesion in the upper pole of right kidney with internal calcific foci [Figure 1].
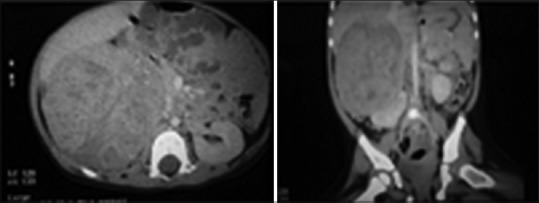
| Figure 1Abdominal computed tomography scan images showing 7.3 cm × 8.0 cm × 9.1 cm heterogeneously enhancing right-sided adrenal tumor
Intraoperatively, the tumor was found adherent to liver, inferior vena cava, and portal vein. Tumor was excised without any spillage. Histopathological examination showed encapsulated tumor mass measuring 9 cm × 8 cm × 7 cm, cut section grayish yellow, soft with areas of hemorrhage and necrosis [Figure 2]. The mass was totally separate from the upper pole of kidney and grossly identified as adrenal mass, the adrenal cortex seen compressed superiorly. The microsections of the tumor showed tumor cells arranged in zellballen and trabeculae pattern. Individual tumor cells show marked variation in size, shape along with spindle cells and finely granular basophilic to amphophilic cytoplasm with intracytoplasmic hyaline globules. Nuclei were round to be oval with prominent nucleoli containing intracytoplasmic inclusions with salt and pepper chromatin at places [Figure 3]. Some of the tumor cells were bizarre and pleomorphic, sparse mitosis with areas of hemorrhage, necrosis, and cystic changes. Lymph nodes did not show any infiltration. Histopathological diagnosis of PCC was documented with advice for urinary vanillylmandelic acid (VMA) estimation. VMA was within normal limit (1.70 [normal 0.00–13.60 mg/24 h]). The child is on follow-up with imaging studies, asymptomatic, and gaining weight.
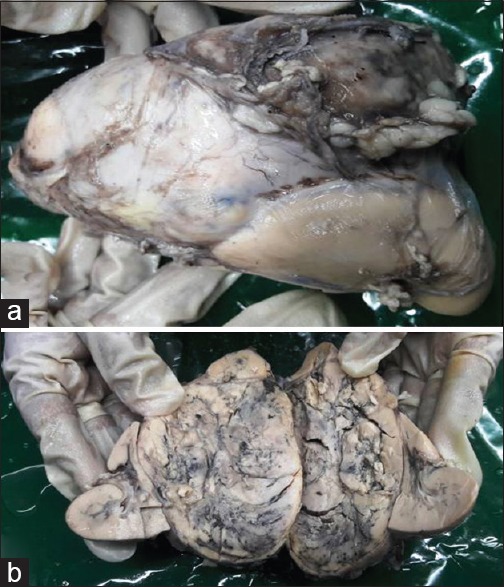
| Figure 2A largxe adrenal medullary mass. (a) Mass on upper pole of kidney. (b) Cut section: Grayish yellow hemorrhagic mass compressing adrenal cortex superiorly and kidney lie on the either sides
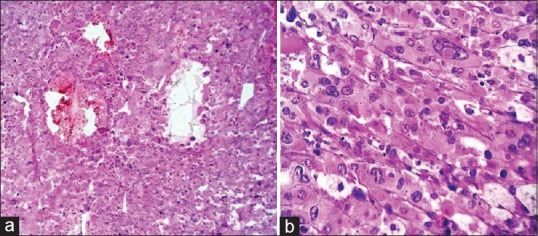
| Figure 3Photomicrograph (a) zellballen nest pattern of tumor cells with hemorrhage and cystic change (H and E, ×100) (b) bizarrely pleomorphic tumor cells with prominent nucleoli and intranuclear inclusions and amphophilic granular cytoplasm (H and E, ×400)
Discussion
Despite its low incidence, PCC is the most common endocrine tumor in children.[1] PHEO is the term used for catecholamine-secreting tumors that occur in the adrenal medulla, the most common location, whereas PGL is extraadrenal tumors that arise from both sympathetic and parasympathetic paraganglia.[2] In a retrospective study of 36 years, Ein et al. published 14 children (10 boys and 4 girls) and found youngest child was 8 years of age.[5] Another series published from Turkey reported 16 children (12 boys and 4 girls) and the mean age at diagnosis was 10.7 years.[6] A Japanese series on PCC published in 2015 revealed the mean age at diagnosis in children as 13.2 years.[1] Our patient was only 14 months of age at diagnosis, and she was symptomatic for the last 1 month. PCC presenting soon after infantile period is an extremely rare finding.
Although rare, PCC comprises 1% of pediatric hypertension.[7] The most common presentation of PCC in children is sustained hypertension. Hypertension is present and sustained in about 90% of affected children at the time of diagnosis.[4] Ciftci et al. detected hypertension in most of their cases as presenting symptom.[6] Hence, PCC without hypertension in a child is very uncommon as in our case. The classic triad of symptoms a headache, palpitation, and diaphoresis recorded in many cases[2] was also not found in our case. The index case presented as a cold case an atypical presentation for childhood PCC.
The diagnosis of adrenal gland phaeochromocytoma is documented here with following features: (1) Characterized by zellballen nests of tumor cells separated by delicate fibrovascular stroma, (2) varying sized and shaped tumor cells with granular cytoplasm and with hyaline globules, (3) nuclear atypia with inclusions, and (4) though marked pleomorphism, necrosis were evident. It is categorized as benign tumor following the principle of all benign endocrine tumors. The evidence of malignancy is not considered in this endocrine tumor as lymph nodes did not show any metastatic deposits. CT scan did not detect any deposit in other organs. Indication for urinary VMA estimation was to exclude the multifocality of the tumor in extraadrenal chromaffin sites which is commonly seen in childhood PCCs. Clinically, another childhood tumor of adrenal medulla named as anaplastic neuroblastoma may come as a differential diagnosis of this tumor because both the tumors show bizarre appearance of the tumor cells and zellballen pattern. Immunohistochemistry showed negative stain which excluded neuroblastoma.
Germ-line mutation of either of six known genes: RET (MEN 2A, 2B), NF1 (Von Recklinghausen neurofibromatosis Type 1), Von Hippel–Lindau syndrome, SDHD, SDHC, and SDHB (mitochondrial succinate dehydrogenase gene mutations associated with PGL) are associated with the familial occurrence of the tumor and those present at young age group as in this case.[7,8] These familial cases are recently recognized as 25% occurrence instead of traditional 10% rule of the past.[8,9] This type of PCC are more often bilateral, but our case has presented unilaterally. Studies show thyroid malignancy in either previous or next generation family members.[10] Family history is not suggestive in this case, hence follow-up is continuing.
Conclusion
PCC is very rarely diagnosed tumor during childhood and being detected in a 14-month-old baby is extremely uncommon. Early diagnosis and complete surgical excision are essential for successful management of these rare children.
PCC is known for its 10% bilaterality, 10% extra-adrenal, 10% malignant, 10% without hypertension, and 10% occurrence in children. PCC unassociated with hypertension and childhood presentation both the features as we observed in our case contribute to the “rule of 10s.”
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Eren E, Saglam H, Caliskan Y, Kiristioglu I, Tarim O. Pediatric patients with pheochromocytoma: Experience of a tertiary health center. Pediatr Int 2015;57:875-9.
- Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, et al. Acurrent review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2010;95:2023-37.
- Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: No longer the 10% tumor. J Surg Oncol 2005;89:193-201.
- Caty MG, Escobar MA Jr. Adrenal tumors. In: Coran AG, Adzick NS, Krummel TM, Laberge J, Shamberger RC, Caldmone AA, editors. Pediatric Surgery. 7th ed. Philadelphia: Elsevier Saunders; 2012. p. 557-66.
- Ein SH, Pullerits J, Creighton R, Balfe JW. Pediatric pheochromocytoma. A 36-year review. Pediatr Surg Int 1997;12:595-8.
- Ciftci AO, Tanyel FC, Senocak ME, Büyükpamukçu N. Pheochromocytoma in children. J Pediatr Surg 2001;36:447-52.
- Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C, et al. Phaeochromocytoma in children. Arch Dis Child 2008;93:899-904.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665-75.
- Bissada NK, Safwat AS, Seyam RM, Al Sobhi S, Hanash KA, Jackson RJ, et al. Pheochromocytoma in children and adolescents: A clinical spectrum. J Pediatr Surg 2008;43:540-3.
- Beck O, Fassbender WJ, Beyer P, Kriener S, Neumann HP, Klingebiel T, et al. Pheochromocytoma in childhood: Implication for further diagnostic procedures. Acta Paediatr 2004;93:1630-4.
| Figure 1Abdominal computed tomography scan images showing 7.3 cm × 8.0 cm × 9.1 cm heterogeneously enhancing right-sided adrenal tumor
| Figure 2A largxe adrenal medullary mass. (a) Mass on upper pole of kidney. (b) Cut section: Grayish yellow hemorrhagic mass compressing adrenal cortex superiorly and kidney lie on the either sides
| Figure 3Photomicrograph (a) zellballen nest pattern of tumor cells with hemorrhage and cystic change (H and E, ×100) (b) bizarrely pleomorphic tumor cells with prominent nucleoli and intranuclear inclusions and amphophilic granular cytoplasm (H and E, ×400)
References
- Eren E, Saglam H, Caliskan Y, Kiristioglu I, Tarim O. Pediatric patients with pheochromocytoma: Experience of a tertiary health center. Pediatr Int 2015;57:875-9.
- Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, et al. Acurrent review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2010;95:2023-37.
- Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: No longer the 10% tumor. J Surg Oncol 2005;89:193-201.
- Caty MG, Escobar MA Jr. Adrenal tumors. In: Coran AG, Adzick NS, Krummel TM, Laberge J, Shamberger RC, Caldmone AA, editors. Pediatric Surgery. 7th ed. Philadelphia: Elsevier Saunders; 2012. p. 557-66.
- Ein SH, Pullerits J, Creighton R, Balfe JW. Pediatric pheochromocytoma. A 36-year review. Pediatr Surg Int 1997;12:595-8.
- Ciftci AO, Tanyel FC, Senocak ME, Büyükpamukçu N. Pheochromocytoma in children. J Pediatr Surg 2001;36:447-52.
- Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C, et al. Phaeochromocytoma in children. Arch Dis Child 2008;93:899-904.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665-75.
- Bissada NK, Safwat AS, Seyam RM, Al Sobhi S, Hanash KA, Jackson RJ, et al. Pheochromocytoma in children and adolescents: A clinical spectrum. J Pediatr Surg 2008;43:540-3.
- Beck O, Fassbender WJ, Beyer P, Kriener S, Neumann HP, Klingebiel T, et al. Pheochromocytoma in childhood: Implication for further diagnostic procedures. Acta Paediatr 2004;93:1630-4.