 PDF
PDF  Views
Views  Share
Share


Paravaginal peripheral primitive neuroectodermal tumor: A rare tumor
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(03): 171-173
DOI: DOI: 10.4103/0971-5851.92823
Abstract
Peripheral primitive neuroectodermal tumor (PNET) of the female genital tract, particularly those in the vaginal and paravaginal region, are extremely rare. A 36-year-old woman presented with clinical features similar to that of a case of cervical fibroid. It was only after surgery that the histopathology suggested it to be a malignant round cell tumour and was CD99 positive. She underwent adjuvant chemotherapy with the Ifosfamide and Etoposide alternating with Vincristine, Doxorubicin, and Cyclophosphamide regime and radical radiotherapy. She is disease free at 12 months of follow-up. The importance of immunostaining and adequate histopathology report lies in the fact that the correct diagnosis thus achieved enabled us to manage a rare case of paravaginal PNET with a multimodality approach.
Publication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Peripheral primitive neuroectodermal tumor (PNET) of the female genital tract, particularly those in the vaginal and paravaginal region, are extremely rare. A 36-year-old woman presented with clinical features similar to that of a case of cervical fibroid. It was only after surgery that the histopathology suggested it to be a malignant round cell tumour and was CD99 positive. She underwent adjuvant chemotherapy with the Ifosfamide and Etoposide alternating with Vincristine, Doxorubicin, and Cyclophosphamide regime and radical radiotherapy. She is disease free at 12 months of follow-up. The importance of immunostaining and adequate histopathology report lies in the fact that the correct diagnosis thus achieved enabled us to manage a rare case of paravaginal PNET with a multimodality approach.
INTRODUCTION
Primitive peripheral neuroectodermal tumors (PNETs) are a part of the spectrum of neoplastic diseases known as the Ewing sarcoma family of tumors ( EFT ) and account for 4% of malignancies. Of this percentage, 15% are of extraskeletal or of soft tissue origin.[1] We present a case of PNET occurring in the paravaginal tissue, which was difficult to diagnose preoperatively. The interesting point here is that this is a rare case and the patient was diagnosed and later treated radically with surgery, chemotherapy, and radiotherapy.
CASE REPORT
The present case is a 36-year-old married multiparous woman, who was suffering from intermittent retention of urine for the last 5 months and later presented with a mass coming out of the vagina for past 3 months. She had no complains of bleeding or discharge from vagina and her bowel habits were normal. Her general physical examination was normal. On detailed gynecologic examination, it was found that there was a fleshy smooth surfaced mass measuring 5 × 4 cm occupying the vaginal cavity. Per speculum examination could not be done owing to lack of space, and on digital palpation a firm mass was felt at the cervicovaginal region on the right side. The overlying vaginal mucosa was smooth. Per rectal examination was normal. A pelvic ultrasonography (USG) revealed a mass in the pelvic region. Chest X-ray, upper abdominal USG, cystoscopy, and proctoscopy yielded normal findings. The overall clinical impression was of a cervicovaginal mass with the provisional diagnosis of a cervical fibroid. Subsequently, she was taken up for surgery. Intraoperative findings revealed a pelvic tumor in the paravaginal region, which was adherent to the surrounding region, and hence was taken out piecemeal. Ultimately, she underwent total abdominal hysterectomy and bilateral salpingo oophorectomy and retroperitoneal lymph node dissection owing to the suspicion of a malignant growth. Postoperative histopathology reported that cervix, vagina, endometrium, tubes, and ovaries were normal. The histology of the tumor was consistent with malignant round cell tumor [Figure 1]. Immunostaining with desmin and cytokeratin was negative but for CD99 it was positive, ultimately favoring the diagnosis of Primitive Neuroectodermal tumor [Figure 2]. The patient underwent adjuvant chemotherapy with three cycles of Ifosfamide and Etoposide alternating with Vincristine, Doxorubicin, and Cyclophosphamide (VAC/IE). After three cycles, she was treated with pelvic radiotherapy to a total dose of 50 Gy in 25 # over 5 weeks. Post radiotherapy she again received three cycles of the chemotherapy. After treatment completion, she had no complains. Her pelvic examination is normal; chest X-ray and whole abdominal and pelvic CT reveals no new lesion or abnormality. She is disease-free 12 months after diagnosis.
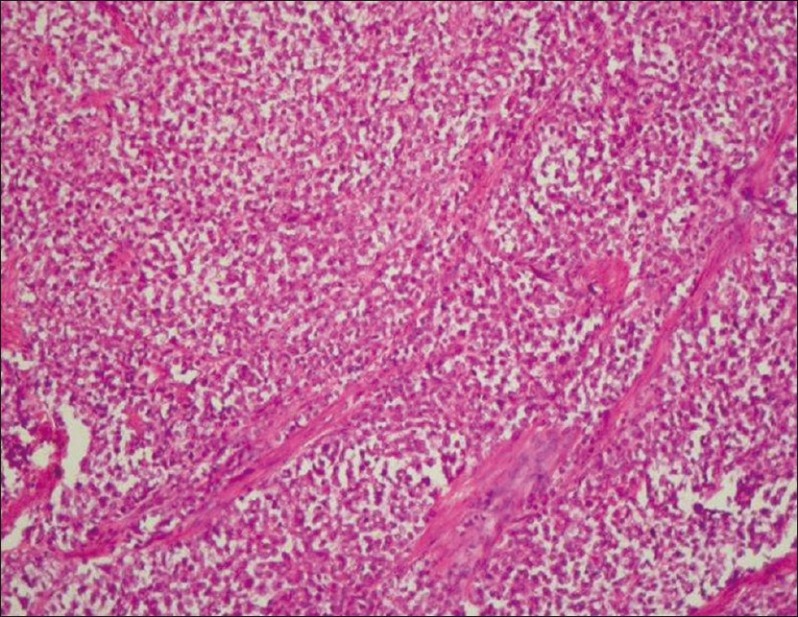
| Fig. 1 Low power view of tumor
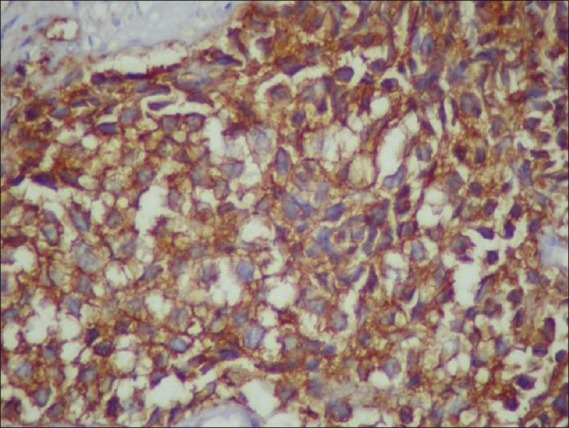
| Fig. 2 CD99-positive cells
DISCUSSION
The PNETs are collectively described as part of the Ewing sarcoma family of tumors. The most common site of PNET in the female genital tract is the ovary. The uterine corpus is the next most common site, with the cervix, vulva, and vagina rarely being the primary site.[2] A review of the literature revealed only seven previously reported possible cases in the vagina, most of which have not had molecular confirmation.[3–5]
The differential diagnosis of vaginal and paravaginal tumors include benign tumors such as cervical fibroids and malignant growths like carcinoma. Among the rare variants of vaginal and paravaginal tumors are melanomas, sarcomas, lymphomas. Lymphomas most closely mimic PNETs with sheets of cytologically malignant small cells without evidence of glandular or squamous differentiation. The absence of epithelial markers like cytokeratin excludes a diagnosis of carcinoma, and to a lesser extent, small cell neuroendocrine carcinomas. Although MIC2 is not specific for Ewing sarcoma/PNET and is expressed in a wide range of unrelated tumors, staining with CD99, as in the present case, provides support for the diagnosis of these tumors, if used with other features.
In early days, management decisions for such tumors revolved around optimal local surgical treatment, followed by additional treatment such as irradiation and/or chemotherapy. More recently, however, neo-adjuvant chemotherapy followed by local treatment has been favored because of improved results by the addition of systemic therapy. The limited prognosis of such patients due to the development of metastatic disease has likewise been improved by using a multimodality treatment and sequencing of different treatment strategies. Owing to its location in the pelvis, the question arises whether radical complete surgery is possible. In case of a purely soft tissue origin, due consideration must be given regarding its extent. The present reported case had a clinical picture quite similar to that of a cervical fibroid. Hence, with the provisional diagnosis of a benign lesion, the patient actually did not undergo any preoperative evaluation regarding the histology, nor she underwent any radiological tests like CT scan to know the extent of the lesion.
However, in our opinion, it indicates that tumor in this location gives symptoms at an early stage. This kind of a presentation though poses a diagnostic dilemma to the clinician but at the same time provokes a surgeon to operate early, thus leading to an early diagnosis postoperatively.
Recent data show that the addition of ifosfamide and etoposide to a standard regimen of vincristine, doxorubicin, and cyclophosphamide significantly improves the outcome for patients with non-metastatic Ewing sarcoma or primitive neuroectodermal tumor.[6] Our patient has now completed radical surgery, adjuvant radiotherapy, and chemotherapy with the regimen of VAC/IE with no evidence of disease 12 months after completion of therapy. Her response supports the use of multimodal therapy for this unusual diagnosis.
The 5-year disease-free survival is 24-80% for localized disease,[7] with smaller resectable lesions; ie, the reported cases with vaginal location had a better prognosis. For other gynecologic locations, such as uterus, survival was related to stage. Two women with stage I disease were alive 5 years after diagnosis; however, two women with stage II and IV tumors died 6 and 12 months after diagnosis, respectively.[8] One patient having disease in the cervix was disease-free 18 months after diagnosis and the other died 4.2 years after diagnosis.[9]
In summary, the present case is the probably the first case reported of PNET of the paravaginal region, and this was possible only because of proper histopathological report and immunostain.
According to studies with these tumors in other locations, they should be treated with adequate surgery followed by adjuvant chemotherapy, similar to that of Ewing sarcoma, and radiation therapy. Prognosis is related to volume of tumor, age at diagnosis, and stage.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Low power view of tumor
| Fig. 2 CD99-positive cells