 PDF
PDF  Views
Views  Share
Share


Myeloproliferative neoplasms working group consensus recommendations for diagnosis and management of primary myelofibrosis, polycythemia vera, and essential thrombocythemia
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2015; 36(01): 3-16
DOI: DOI: 10.4103/0971-5851.151770
E X E C U T I V E S U M M A R Y
According to the 2008 revision of the World Health Organization (WHO) classification of myeloid malignancies, philadelphia chromosome (Ph)-negative myeloproliferative neoplasms (MPNs) include clonal, hematologic disorders such as polycythemia vera, primary myelofibrosis, and essential thrombocythemia. Recent years have witnessed major advances in the understanding of the molecular pathophysiology of these rare subgroups of chronic, myeloproliferative disorders. Identification of somatic mutations in genes associated with pathogenesis and evolution of these myeloproliferative conditions (Janus Kinase 2; myeloproliferative leukemia virus gene; calreticulin) led to substantial changes in the international guidelines for diagnosis and treatment of Ph-negative MPN during the last few years. The MPN-Working Group (MPN-WG), a panel of hematologists with expertise in MPN diagnosis and treatment from various parts of India, examined applicability of this latest clinical and scientific evidence in the context of hematology practice in India. This manuscript summarizes the consensus recommendations formulated by the MPN-WG that can be followed as a guideline for management of patients with Ph-negative MPN in the context of clinical practice in India.
Keywords
Essential thrombocythemia - myeloproliferative neoplasms - polycythemia vera - primary myelofibrosisPublication History
Article published online:
12 July 2021
© 2015. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
- According to the 2008 revision of the World Health Organization (WHO) classification of myeloid malignancies, philadelphia chromosome (Ph)-negative myeloproliferative neoplasms (MPNs) include clonal, hematologic disorders such as polycythemia vera, primary myelofibrosis, and essential thrombocythemia.
- Recent years have witnessed major advances in the understanding of the molecular pathophysiology of these rare subgroups of chronic, myeloproliferative disorders. Identification of somatic mutations in genes associated with pathogenesis and evolution of these myeloproliferative conditions (Janus Kinase 2; myeloproliferative leukemia virus gene; calreticulin) led to substantial changes in the international guidelines for diagnosis and treatment of Ph-negative MPN during the last few years.
- The MPN-Working Group (MPN-WG), a panel of hematologists with expertise in MPN diagnosis and treatment from various parts of India, examined applicability of this latest clinical and scientific evidence in the context of hematology practice in India.
- This manuscript summarizes the consensus recommendations formulated by the MPN-WG that can be followed as a guideline for management of patients with Ph-negative MPN in the context of clinical practice in India.
INTRODUCTION
The philadelphia chromosome (Ph)-or bcr-abl negative myeloproliferative neoplasms (MPNs) represent a range of clonal hematological diseases with overlapping clinicopathological features. The main entities, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are characterized by clonal excess hematopoiesis in one or more cell lines.[1] Both PV and ET can progress to myelofibrosis (MF), and all three entities can transform into acute myeloid leukemia (AML).[2,3] The incidence rates for classic, Ph-negative MPNs fall in the range of 0.1-2.8 cases/100,000/year in United States and European Union where systematic registry studies are available. Estimated incidence rates are higher for PV (0.4-2.8/100,000/year) compared to ET (0.38-1.7/100,000/year) and MF (0.1-1/100,000/year) based on data from various registries from European Union. According to a recent study based on US registries, prevalence rates are higher for PV (44-57/100,000) and ET (38-57/100,000) compared to MF (4-6/100,000). This may be due to longer median survival in PV and ET (8-10 years) compared to MF (2-5 years).[4,5] Specific studies addressing incidence and prevalence of Ph-negative MPNs are not available in the context of developing countries like India.
Recent advances in the understanding of the molecular pathogenesis of Ph-negative MPNs offer scope for improving diagnosis of these rare hematologic malignancies with the help of universally applicable simple tests. In 2005, many research groups simultaneously identified mutations in Janus Kinase 2 (JAK2), a protein tyrosine kinase playing a central role in cytokine signal transduction, as the most frequent molecular abnormality in PV (~90% of cases), ET and MF (~50% of cases). These findings have modified the landscape of diagnosis, risk stratification, treatment and response assessment in Ph-negative MPNs.[6,7,8] The MPN Working Group (MPN-WG) has discussed how these latest developments can be applied to the context of hematology practice in India and formulated consensus recommendations to improve diagnosis and prognostic stratification of patients with Ph-negative MPNs.
Traditional treatment approaches for PV and ET focus on prevention of thrombotic events with aspirin along with antiproliferative agents. Treatment strategies for MF are driven by the clinical presentations like anemia and/or splenomegaly. Few randomized controlled trials are available in the field of Ph-negative MPN to guide decision-making for individual patients. In clinical practice, many issues remain uncertain, demanding high degree of professional experience. Improved understanding of the molecular pathophysiology of these entities in recent years has prompted an investigation of targeted treatments in MF and PV. This article summarizes extensive discussions held during the MPN-WG meetings regarding relevance and applicability of these new treatment options in the Indian scenario. Based on these discussions, the MPN-WG has formulated recommendations for diagnosis and treatment of Ph-negative MPNs in order to assist clinicians in optimizing patient care.
METHODS
The MPN-WG is a panel of expert physicians from India, selected for their expertise in research and clinical management of Ph-negative classical MPNs. The panel met twice during November 2013 and February 2014 with the objective of formulating the consensus recommendations for diagnosis and management of Ph-negative MPNs along with various other objectives including raising awareness about MPNs through continuing medical education programs, conducting pathology workshops to improve diagnosis and collaborating on MPN related research. In order to draft the recommendations for Ph-negative classical MPNs, the panel formed two committees; one for diagnosis and the other for management of Ph-negative MPNs. The panel members searched databases like PubMed for available literature on Ph-negative classical MPNs for formulating the guidelines. For each recommendation, references from international guidelines are provided for the level of evidence. As there is limited information available on treatment practices in the Indian context, some of the recommendations are based on the experience of the authors. The draft prepared by each committee was circulated to all the members of the group for their critical feedback. Subsequently, the group met face-to-face to have a final discussion on each subject and formulate the consensus recommendations. Evidence levels and recommendation grades are as per [Appendix 1].[9]
Diagnostic criteria for Ph-negative classical myeloproliferative neoplasms
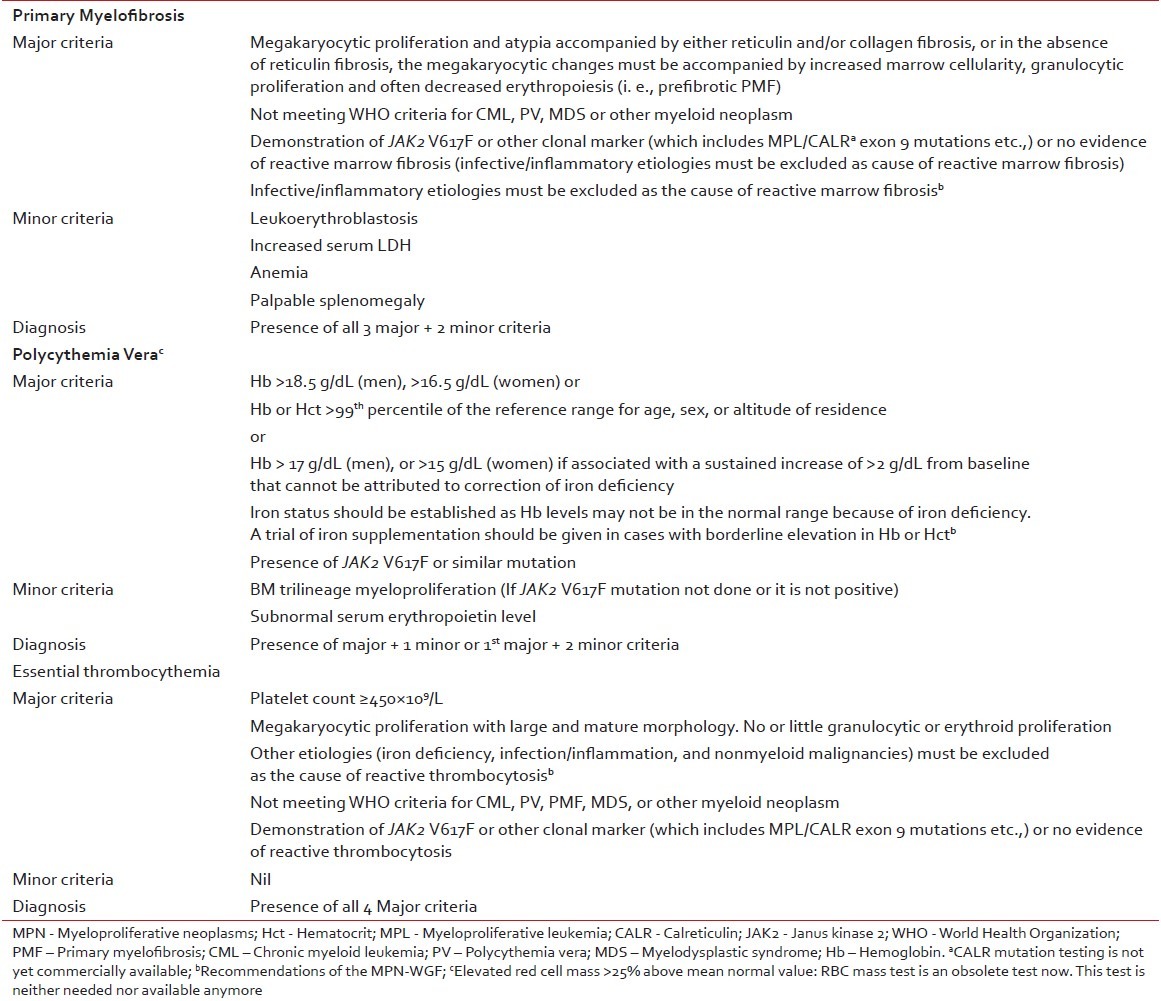
World Health Organization (WHO) 2008 classification and diagnosis criteria for Ph-negative MPNs have been widely accepted as standardized, uniform diagnostic criteria to be followed for clinical research, case reporting, and clinical practice. The panel agreed that these criteria can be adapted in the Indian context with a few additional measures [Table 1]. Even though red cell mass measurement has been cited as a valid diagnostic tool for PV in the WHO 2008 diagnostic criteria, this test is not commonly available. In the context of hematology practice in India, diagnostic workup for MPNs should be performed taking into account the fact that hemoglobin levels may not be in the normal range due to nutritional deficiencies. When marginal hemoglobin deficiency is observed, common nutritional causes should be diagnosed by simple blood tests and corrective measures should be taken such as iron supplementation in patients where required. Application of morphology criteria to distinguish ET from prefibrotic PMF will be challenging in India due to shortage of specialized centers. WHO 2008 classification system for Ph-negative MPNs addresses this issue by incorporating biologically relevant minor criteria to confirm histologic pattern indicative of prefibrotic PMF.[10] The panel suggested that pathology slides should be referred to MPN reference centers to establish confirmed diagnosis.
Table 1
Diagnostic criteria for MPN (adapted from WHO 2008 diagnostic criteria)
Baseline workup for patients with Ph-negative classical myeloproliferative neoplasms
The panel reached a consensus regarding baseline diagnostic workup to be followed in the case of suspected Ph-negative MPN [Figure 1]. General physical examination, biochemical and pathological assessments, cytogenetics, and mutation analysis should be done for all patients with Ph-negative classical MPNs. Molecular testing for JAK2 V617F mutation in peripheral blood sample by polymerase chain reaction can be incorporated in the baseline workup to guide risk-adapted therapy in patients with Ph-negative classical MPNs. In JAK2 V617F negative patients, myeloproliferative leukemia and calreticulin mutation testing can be performed to assist further in diagnosis and to exclude other etiologies as cause of reactive thrombocytosis.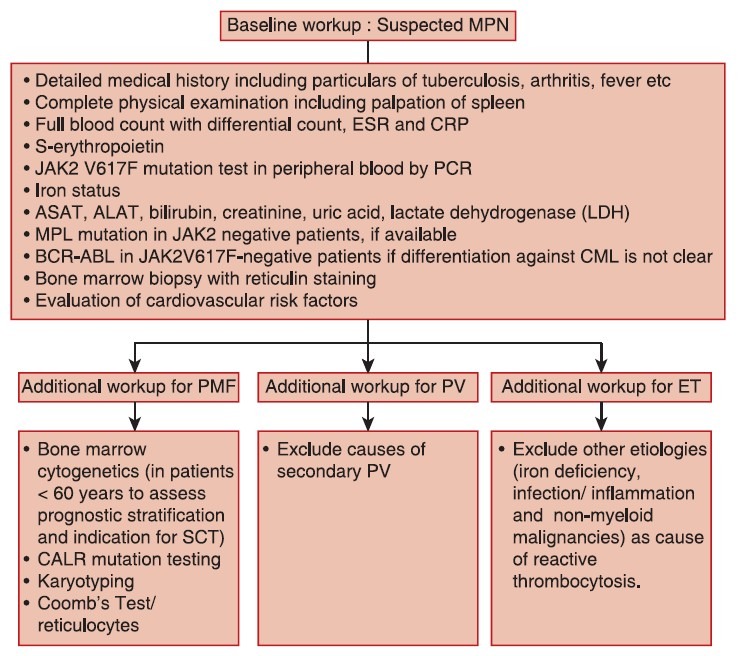
| Fig. 1 Baseline workup for classical philadelphia chromosomenegative myeloproliferative neoplasms
Primary myelofibrosis
Treatment goals
Treatment strategies in patients with PMF aim at improving survival, possible cure in patients eligible for allogeneic stem-cell transplantation (allo-SCT), minimizing risk of transformation to acute leukemia, avoiding first occurrence or recurrence of thrombotic and bleeding complications, treating anemia and other cytopenias, managing symptomatic splenomegaly, and treating symptoms (weight loss, night sweats, fever, pruritus, fatigue etc.). Potential high-risk situations such as surgery should be anticipated and well-managed. Therapy should be directed toward improving quality-of-life.[9,10]
Risk stratification
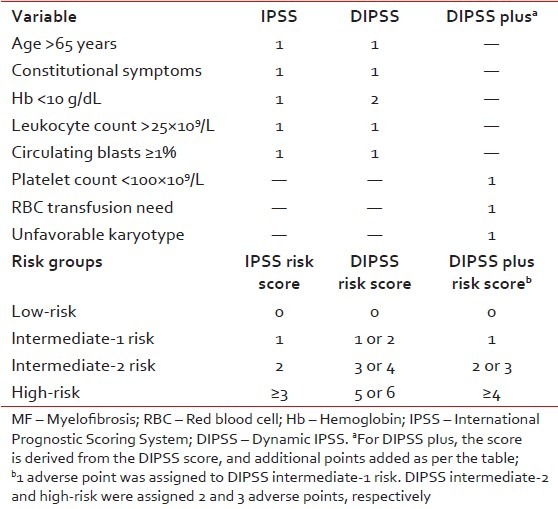
Treatment decisions in PMF are often challenging, particularly with regards to timing of allo-SCT or participation in clinical trials. Therefore, accurate risk stratification of patients in terms of overall and leukemia-free survival is critical. In this regard, survival from the time of diagnosis is best assessed by the International Prognostic Scoring System (IPSS)[11] whereas a dynamic IPSS (DIPSS) model is used for estimating survival at any point in the disease course [Table 2].[12] Both IPSS and DIPSS utilize the same five risk factors for survival (age >65 years, hemoglobin < 10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, and constitutional symptoms) in order to classify patients into four risk groups: Low, intermediate-1 (Int-1), Int-2, and high-risk. More recently, IPSS-independent prognostic factors for survival in PMF have been described and include red cell transfusion need, unfavorable karyotype, and thrombocytopenia.[13] The DIPSS plus effectively combines prognostic information from DIPSS, karyotype, platelet count, and transfusion status to predict overall survival in PMF.[13]
Table 2
Risk stratification for patients with MF
Clinical management
After reviewing the available evidence for treatment outcome in controlled trials, the panel reached a consensus that clinical management of PMF should combine available treatment options directed toward the control of anemia, thromobocytopenia, and splenomegaly. As a result of these discussions, a comprehensive treatment algorithm was compiled for PMF, integrating traditional and innovative targeted treatment strategies [Figure 2].
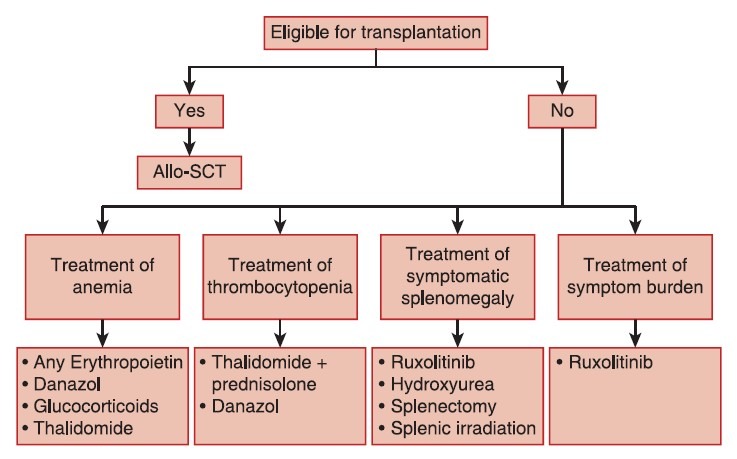
| Fig. 2 Treatment algorithm for primary myelofibrosis
Treatment of anemia
As a general guideline, pharmacological treatment of anemia should be initiated at hemoglobin levels approximately < 11 g/dL in symptomatic patients, and should be considered in asymptomatic patients with hemoglobin levels < 10 g/dL.[9,10] Anemia in PMF is multifactorial and deficiency of iron, Vitamin B12, and folic acid should always be ruled out and corrected before considering other therapies. Furthermore, reticulocyte count and Coomb's test should be performed to rule out immunohemolytic conditions. Since baseline reticulocyte count can be higher in patients with PMF, these results need to be interpreted with caution.
Erythropoietin
Erythropoietin (EPO) levels should be measured before starting therapy with recombinant human EPO.
If EPO levels are < 500 IU, then EPO replacement therapy can be considered. The starting dose of EPO is 10,000 U once weekly. The response should be monitored for 2 weeks, and if no response is observed, the dose can be escalated up to 40,000 IU once weekly. If no further response is seen after the maximum recommended dose for 8-12 weeks of EPO, therapy should be discontinued. Darbepoietin-α administered bi-monthly is equally effective, and the recommended dose is in the range 150-300 μg/fortnight. The goal is to maintain hemoglobin level at approximately 12 g/dL (11 g/dL for females) in patients with PMF. Recombinant human EPO should be stopped, or the dose reduced when hemoglobin is above 12 g/dL to avoid risk of thrombosis.[9]
Danazol
Danazol is administered at a dose of 200 mg twice daily. Monitoring of liver function is regularly recommended (once monthly). Most patients respond within the first 2-3 months. The therapy should be discontinued if no response is observed after a 4 months trial of these agents. A synergistic effect between human recombinant EPO and danazol treatment has been reported.[9,15]
Glucocorticoids (mostly used in combination with thalidomide)
Glucocorticoid therapy is recommended in patients with hemolytic activity but may also be useful at dose of 10-20 mg/day in some patients with anemia without hemolytic activity. In patients with hemolysis and a positive Coomb's test, an initial test dose of 1 mg/kg is recommended. Grade C recommendation, evidence level IV.[9]
Thalidomide
Thalidomide can increase the hemoglobin level and decrease spleen size in PMF patients. Low-dose thalidomide (50 mg/day preferably given at bedtime) in combination with prednisolone can improve anemia in 20-30% of patients.[16] However, thalidomide is associated with nonhematological toxicity (constipation, sedation, depression, peripheral neuropathy), and is contraindicated in pregnancy. It is to be used with caution and effective contraception in women of child-bearing age.
Recommendation
Low-dose thalidomide (maximum 50 mg/day) in combination with prednisolone (10-20 mg/day for 2 weeks and afterward tapering to the lowest dose necessary for maintaining an adequate hemoglobin-concentration) can be considered for patients not responding to EPO or danazol. Therapy should be discontinued if no response is seen after 4 months. Grade B recommendation, evidence level III.[9]
The panel agreed that standard transfusion guidelines should be applied for patients eligible for transfusion to correct anemia associated with MF.
Treatment of thrombocytopenia
The panel has examined the available evidence for treatment of thrombocytopenia associated with MF and concluded that in the context of hematology practice in India, thalidomide in combination with prednisolone should be the choice of therapy.[17] If no response is observed with this combination, danazol is recommended.
Treatment of symptomatic splenomegaly
Hydroxyurea
The efficacy and safety of hydroxyurea (0.5-2 g/d) in the treatment of PMF have been reported in very few studies with relatively small number of patients.[18,19]
Recommendation
Hydroxyurea is recommended as cytoreductive therapy in older PMF patients not eligible for transplantation. Grade B recommendation, evidence level III.[9]
Janus kinase 2 inhibitors
Several JAK2 inhibitors have been tested, but until now ruxolitinib is the only approved drug available. Ruxolitinib has been shown to reduce spleen volume by at least 35% in 40% of patients with Int-2 or high-risk disease in two large, randomized controlled trials; and 50% reduction in spleen length in 50%, 15%, and 48% of patients with Int-1, Int-2 and high-risk disease, respectively in a phase 2 trial.[20,21,22] So far, there is no clear evidence that ruxolitinib can slow disease progression. However, a survival benefit has been reported in patients on ruxolitinib when compared to patients on placebo or best available therapy.[20,21] Ruxolitinib is the current treatment of choice for constitutional prognostic symptoms of the disease (weight loss, fever, and night sweats), spleen related symptoms (such as abdominal discomfort, pain under left ribs etc.), and disease-related symptoms (such as itching, fatigue) as no other therapy has been shown to significantly improve symptom burden and quality-of-life in MF.[23] A common misconception is that JAK inhibition is effective primarily in patients who have the JAK2 V617F mutation. However, ruxolitinib demonstrated comparable efficacy in patients with or without the V617F mutation. Since JAK2 is involved in thrombopoietin and EPO signaling, dose-dependent thrombocytopenia and anemia should be anticipated and managed appropriately in MF patients undergoing treatment with JAK2 inhibitors such as ruxolitinib. Starting dose of ruxolitinib should be personalized according to baseline platelet counts [Table 3]. In MF patients with baseline platelet counts between 100 and 200 × 109/L, ruxolitinib starting dose of 15 mg twice daily is recommended along with frequent monitoring of platelet and absolute neutrophil counts.[24,25,26]
Table 3
Ruxolitinib dose in primary MF
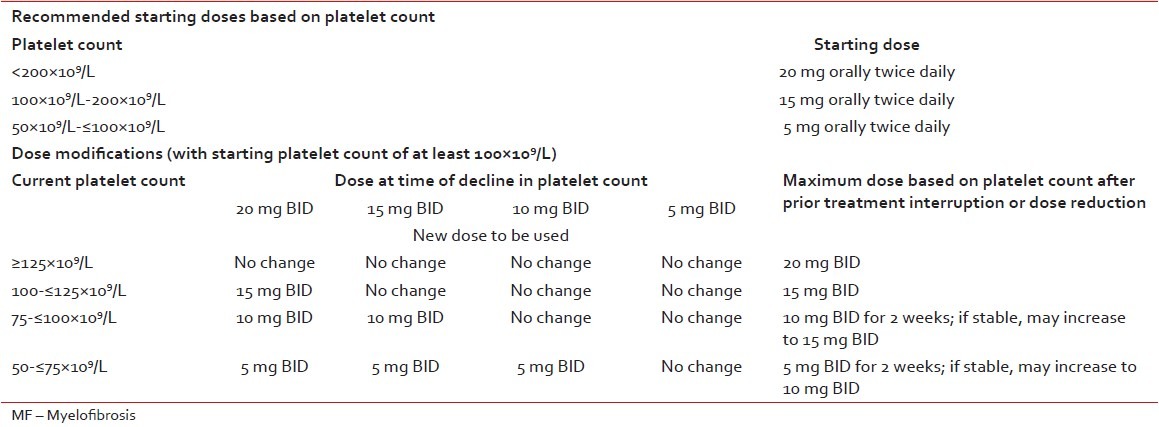
Recommendation
Ruxolitinib should be considered as first-choice of treatment in patients with symptomatic splenomegaly [Table 3].[22,23,24,25,26] Grade A recommendation, evidence level 1b.[9]
Splenectomy
Even though splenectomy procedure is associated with significant morbidity (25-30%) and mortality (7-10%), in a restricted subgroup of MF patients with refractory splenomegaly, this option should be carefully evaluated. Significant surgical expertise, fastidious surgical hemostasis, and careful control of postoperative thrombocytosis have been found to be essential in trying to minimize the risk of this procedure. Critical factors to be assessed before splenectomy include type of conditioning used, timing of the intervention and expertise of the surgeon.[27]
Recommendation
Splenectomy should not be considered for symptomatic splenomegaly. Splenectomy should be considered only in patients who are not responsive to hydroxyurea, interferon-α, and/or ruxolitinib, with marked splenomegaly associated complications such as splenic infarct, splenic abscess, or rupture, repeated upper gastrointestinal bleeding episodes due to portal hypertension and/or cytopenias secondary to hypersplenism. Grade B recommendation, evidence level III.[9]
Splenic irradiation
Splenic irradiation should be reserved for patients in the indications mentioned above who are not responsive to conventional and novel therapies and patients not eligible for splenectomy. Grade B recommendation, evidence level III.[9] Splenic embolization is not recommended for symptomatic splenomegaly in MF.
Treatment of symptom burden
Ruxolitinib is the current treatment of choice as no other therapy has been shown to significantly improve symptom burden and quality-of-life in MF. Grade A recommendation, evidence level 1b.[9]
Combination therapy
As further details of molecular mechanisms leading to Ph-negative MPNs emerge, combinations of targeted agents are under investigation to improve outcomes in these patients. In preclinical studies, pan-histone deacetylase inhibitor panobinostat has shown a synergistic effect when combined with ruxolitinib especially due to deacetylation of protein chaperone heat shock protein 90 involved in JAK-signal transducers and activators of transcription signaling.[28] Preliminary results from a Phase1b study in MF patients have provided supportive evidence for combination treatment strategies in MF.[29] However, such therapy should only be considered in the clinical trial setting.
Stem-cell transplantation
Since allo-SCT is the only curative treatment in PMF, this option should be considered in all PMF patients at the time of diagnosis. It is recommended in eligible patients (no/minimal associated comorbidities, younger age, availability of HLA-matched donor) with Int-2 or high-risk MPN at diagnosis, and during follow-up of younger low/Int-1 patients that progress to a higher-risk using DIPSS or DIPSS plus.[11,12,13] Outcome is better for patients with low-risk disease, but due to the high procedure-related morbidity and mortality, transplants should only be performed in patients with an expected survival of < 5 years which includes patients with IPSS, DIPSS, or DIPSS plus risk score of Int-2 or high-risk.[11,12,13,30] Patients above 45 years have a very poor survival on myeloablative conditioning.[31]
Recommendation
Allogeneic stem-cell transplant with myeloablative or reduced-intensity conditioning is indicated in young ( < 40 years of age), Int-2 or high-risk patients with PMF. Reduced intensity transplantation should be considered for patients aged 40-60 years with Int-2 or high-risk at diagnosis or later during the course of the disease. Grade B recommendation, evidence level III.[9]
Avoiding thrombotic and bleeding complications
Retrospective analyses indicate that the incidence of thrombotic complications was similar in PMF and ET.[32,33] No prospective trials of platelet reducing agents or aspirin have been performed in PMF. The panel suggested that clinicians should follow the guidelines provided for ET regarding thrombosis and bleeding prevention in PMF. Other factors contributing to atherosclerosis should be addressed. General care guidelines for preventing thrombosis should be followed.
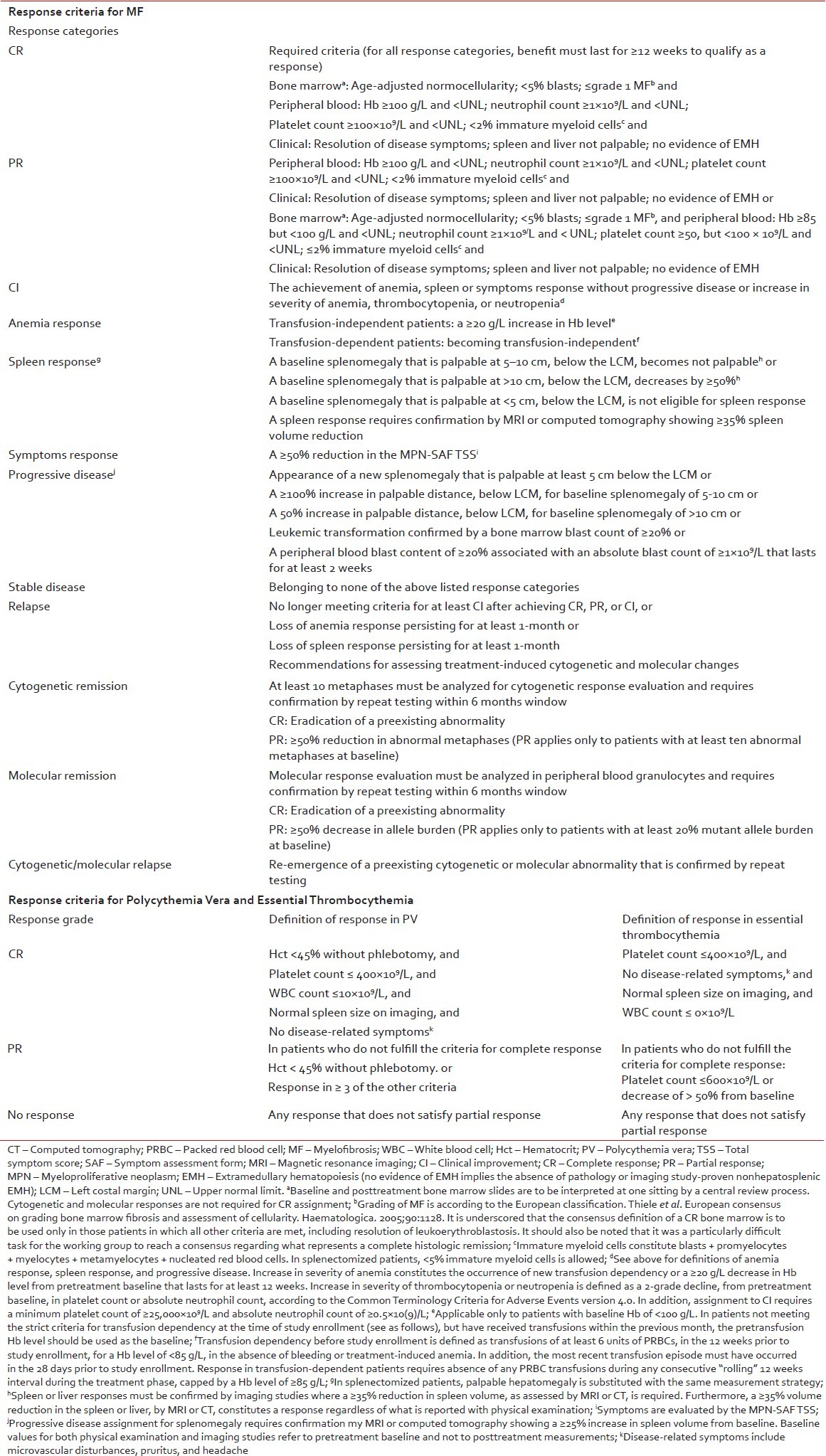
Evaluation of response and follow-up
Table 4
Definition of response in Philadelphia chromosome-negative MPNs
Polycythemia vera
Treatment goals
Major treatment objectives in patients with PV include minimizing the risk of thrombosis and progression; reducing constitutional symptoms, maintaining hematocrit < 0.45, and managing special situations like pregnancy or cardiovascular risk.
Risk stratification
Current risk stratification strategies in PV are designed to estimate the likelihood of thrombotic complications and do not necessarily estimate survival or risk of leukemic/fibrotic transformation.[36] High-risk is defined by age above 60 years, previous thrombosis or platelets more than 1500 × 109/L. These patients should be treated with cytoreductive therapy. Low-risk is defined by the absence of these risk factors and patients in general, should not receive cytoreductive therapy.
Clinical management
The panel examined clinical evidence for traditional treatment approaches in PV and recommendations were formulated based on the consensus. The treatment algorithm for PV is provided in Figure 3.
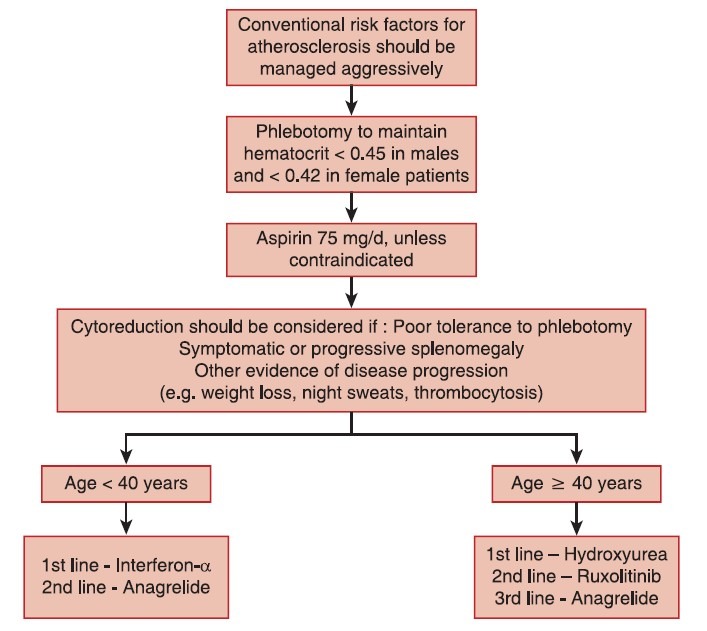
| Fig. 3 Treatment algorithm for polycythemia vera
Phlebotomy
The hematocrit should be maintained at < 0.45 and < 0.42 in male and female patients, respectively.[37] Phlebotomy is recommended for hematocrit levels >0.48. Grade A recommendation, evidence Level Ib.[9]
Aspirin therapy
Aspirin has been shown to reduce both arterial and venous thrombosis in PV.[38] Aspirin should not be given to patients with platelets more than 1,500 × 109/L due to an increased risk of bleeding, instead cytoreductive therapy should be initiated. In the case of aspirin allergy, clopidogrel can be used. Grade A recommendation, evidence level Ib.[9]
Choice of cytoreductive therapy in polycythemia vera
Interferon -α
Interferon-α can be considered in younger patients ( < 40 years). For patients >40 years of age, hydroxyurea should be considered as the first line of treatment. Grade B recommendation, evidence level III.[9]
Hydroxyurea
Hydroxyurea is the best-documented drug in PV having been used in large randomized trials.[39,40] Hydroxyurea is recommended as a first-line cytoreductive therapy in PV patients >60 years or younger patients who do not tolerate interferon-α (Grade A recommendation, evidence level Ib). Increased risk of leukemic transformation has not been observed despite long-term hydroxyurea treatment. Hydroxyurea should be continued till patients develop intolerance or progression. Grade A recommendation, evidence level Ib.[9]
Janus kinase 2 inhibitors
Ruxolitinib provided rapid and durable clinical benefits in patients with advanced PV who were refractory or intolerant to hydroxyurea in a phase 2 clinical trial.[41] These findings were recently confirmed in a phase 3 trial in hydroxyurea refractory or intolerant PV patients that compared 10 mg twice daily ruxolitinib treatment to best available therapy.[42] However, ruxolitinib is presently not approved for use in PV and should not be used outside of the context of clinical trials.
Anagrelide
Anagrelide has powerful platelet reducing activity that could be helpful in the management of patients’ intolerant or refractory to hydroxyurea, interferon-α, or ruxolitinib. Grade C recommendation, evidence level IV.[9]
Radioactive phosphorus
Due to its leukemogenic effect, radioactive phosphorus is not a recommended option for patients < 75 years.[9]
Evaluation of response and follow-up
The goal of therapy should be normalization of peripheral blood counts and the ELN criteria for PV should be used for evaluation of response [Table 4].[43] Patients on phlebotomy alone should be monitored with complete blood counts every 4-6 weeks. There is no indication for repeated bone marrow trephine biopsies during routine follow-up in PV but is essential in assessing the transformation to MF or acute leukemia. Monitoring of molecular response, including sequential assessment of the JAK2 V617F allele burden is at the moment not recommended for routine clinical use.
Essential thrombocythemia
Treatment goals
Similar to PV, main treatment goals in ET include the minimizing risk for thrombosis and progression, normalizing peripheral blood counts, reducing constitutional symptoms, and managing special situations like pregnancy.
Risk stratification
Risk stratification in ET is based on the assessment of risk of thrombosis, as the current therapy in ET is aimed at lowering the risk of thrombosis. True ET diagnosed according to the 2008 WHO classification has not been reported to affect the life expectancy of patients.[32] High-risk is defined as the presence of age above 60 years or history of previous thrombosis, or a platelet count more than 1,500 × 109/L; and these patients should be treated with cytoreductive therapy.[9] Low-risk is defined by the absence of these 3 factors and should not be treated with cytoreductive therapy except in patients with uncontrolled cardiovascular risk factors.[9]
Clinical management
Patients with ET are at high-risk for thrombosis. Hence, vigorous treatment is required for managing cardiovascular risk factors. It is important to emphasize that before starting therapy patients should be evaluated for eventual progression to MF, if they show symptomatic or progressive splenomegaly, other evidence of disease progression such as weight loss, night sweats, progressive leukocytosis, and/or thrombocytosis.
Stem-cell transplantation is almost never performed in ET due to unfavorable risk-benefit profile. The treatment algorithm for ET is provided in Figure 4.
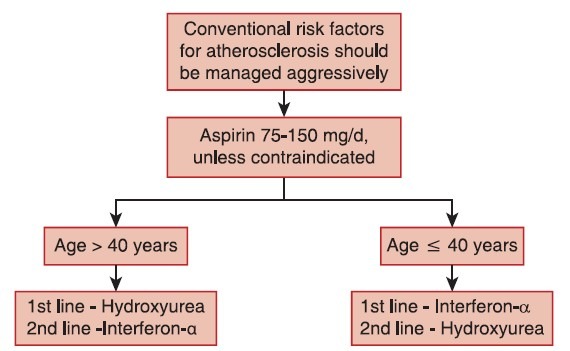
| Fig. 4 Treatment algorithm for essential thrombocythemia
Aspirin
Antiplatelet therapy with aspirin 75-150 mg/day is recommended, unless otherwise contraindicated, for all ET patients.
Choice of cytoreductive therapy in essential thrombocythemia
Hydroxyurea
Hydroxyurea is the best-documented therapy in ET and is recommended as a first-line therapy in the majority of ET patients. Hydroxyurea markedly reduces thrombotic complications compared to aspirin alone.[44]
Interferon-α
Interferon-α treatment is well-documented and safe in ET and is not considered leukemogenic or teratogenic.[43]
Recommendation
Interferon-α is the recommended first-line therapy in younger patients. It can be used in older patients if long-term use of hydroxyurea is not suitable and in patients who do not tolerate hydroxyurea (Grade B recommendation, evidence level III).[9] Interferon-α is the treatment of choice if cytoreductive therapy is indicated during pregnancy or when pregnancy is planned.
Janus kinase 2 inhibitors
Since no JAK2 inhibitor has been studied extensively in ET to date, these drugs are at the present time experimental and should not be used outside of the context of clinical trials.
Other drugs
Other drugs, such as busulfan and radioactive phosphorus, are not relevant in ET. Anagrelide has many complications and may not be as effective as hydroxyurea, but may be used in some patients, if necessary, in combination with hydroxyurea to control platelet counts.
Response monitoring in essential thrombocythemia
The goal of therapy should be to normalize peripheral blood counts in patients who can tolerate pharmacological intervention. The ELN response criteria for ET should be used for evaluation of response [Table 4].[43] There is currently no absolute evidence for a correlation between platelet levels < 400 × 109/L and the reduced risk of thrombosis. Therefore, in patients who develop anemia on hydroxyurea treatment, lowering the dose to allow for higher platelet number in order to avoid anemia is acceptable. There is no indication for repeated bone marrow trephine biopsies in routine follow-up in ET but is essential in assessing the transformation to MF or acute leukemia. Monitoring of molecular response, including sequential assessment of the JAK2 V617F allele burden, is at the moment not recommended for clinical use.
Management of complications of myeloproliferative neoplasms
Acute thrombotic events and secondary prophylaxis
In general, acute thrombotic events should be treated as in non-MPN patients. Control of hematocrit and platelet count should be optimized. In emergency situations such as acute cerebrovascular complications or severe digital ischemia, acute platelet apheresis or erythropheresis can be used in order to achieve a rapid reduction in blood counts. Since the effect is brief, cytoreductive therapy, preferably with hydroxyurea should be started as soon as possible in patients, not on cytoreductive therapy. Prevention of re-thrombosis should be independently achieved in patients with previous venous thrombosis by both oral anticoagulants and antiplatelet drugs. Since no prospective trials exist, it remains unclear whether it is better to give a short course of warfarin or to continue with long-term therapy for secondary prevention of venous thromboembolism.
Bleeding
The most important cause of bleeding in ET and PV is acquired von Willebrand΄s syndrome associated with high platelet counts (>1,500 × 109/L).[45,46] Therefore, the most important therapeutic intervention to manage acute bleeding in the thrombocythemic patient is platelet reduction, and the recommended agent is hydroxyurea.
Platelet apheresis is indicated when extreme thrombocytosis is accompanied by an urgent need to reduce platelet counts, that is, severe or life-threatening bleeding.[47]
Pruritus
Pruritus, typically aquagenic, can be a severe clinical problem in PV. Antihistamines may be of benefit. Selective serotonin re-uptake inhibitors can also lead to improvement of pruritus. Benefit has been shown with phototherapy using psoralen and ultraviolet A light.
Transformation to acute myeloid leukemia
The results after conventional AML induction chemotherapy are dismal in patients developing AML after PV, ET, or PMF, with a very short median survival.[48] If possible, it is recommended that patients undergo allo-SCT after induction chemotherapy. Hypomethylating agents can be used as a bridge to transplant.[49,50]
Special issues
Pregnancy
There is limited information in the medical literature about the management of MPNs in pregnancy.[51] The live birth rate is about 60% due to an overall incidence of first trimester miscarriage of 31-36% (about twice the normal rate) and an increased risk of intrauterine growth retardation, intrauterine death, and stillbirth (8%). Major maternal complications are less common and occur in approximately 8% of ET patients.[52]
Pregnancy is likely to be accompanied by a high-risk of complication for the mother and/or fetus if any of the following factors are present: Previous venous or arterial thrombosis in mother, previous hemorrhage attributed to PV/ET, previous pregnancy complication that may have been caused by PV/ET (these include significant ante-or postpartum hemorrhage, severe preeclampsia, unexplained recurrent first trimester loss (≥3), intrauterine growth retardation (< 5% for gestation), intrauterine death or stillbirth with no other cause identified, placental abruption), and platelet count above 1,000 × 109/L.[53]
Therapeutic options include antithrombotic treatment, phlebotomy in PV and cytoreductive agents. The optimal hematocrit in pregnancy has yet to be established, but the current recommendation is to maintain the hematocrit within the normal range appropriate for gestation. The increased plasma volume often results in a reduced hematocrit and platelet count during the second trimester. The levels rise again during the postpartum period contributing to an increased thrombotic risk during the first 6 weeks after delivery. Close monitoring of blood counts is important during this period.[53] Low dose aspirin is safe and seems advantageous during pregnancy in ET. Starting on the day of the delivery, aspirin is substituted by a prophylactic dose of low molecular weight heparin (LMWH) which is given until 6 weeks after delivery.[51] The doses of LMWH that have been reported are dalteparin 5000 U or enoxaparin 40 mg daily.
Pediatric myeloproliferative neoplasm
The incidence of different MPN in patients aged < 20 years is so low that formal evidence-based recommendations are impossible to provide.
DISCUSSION
Limited information is available from controlled, randomized clinical trials for the management of Ph-negative MPNs and consequently, clinical decision making is extremely challenging. The recommendations presented in this manuscript are mainly based on the clinical experience and knowledge of experts in the field of Ph-negative classical MPNs and a group decision making process was adopted while interpreting available clinical evidence in the context of hematology practice in India. Wherever possible, an expert panel has provided practical suggestions to assist clinicians in diagnosing and treating patients with PMF, PV, or ET. The goals of current therapy for Ph-negative classical MPNs are to address major clinical issues in PMF and to prevent the risk of thrombosis in PV and ET. The challenges in diagnosis are addressed by the recently revised WHO classification, which integrates hematologic, morphologic, and molecular parameters to separate the three clinical entities. Better understanding of molecular mechanisms contributing to the pathogenesis and progression of Ph-negative MPNs during the recent years will translate into more accurate diagnosis and targeted treatment approaches in the near future. It is the intention of this group to update periodically and modify this consensus statement as and when more data become available.
Appendix 1: Evidence levels and recommendations grades[9]

Appendix 2: Myeloproliferative neoplasm symptom assessment form total symptom score[35]
.jpg)
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Baseline workup for classical philadelphia chromosomenegative myeloproliferative neoplasms
| Fig. 2 Treatment algorithm for primary myelofibrosis
| Fig. 3 Treatment algorithm for polycythemia vera
| Fig. 4 Treatment algorithm for essential thrombocythemia