 PDF
PDF  Views
Views  Share
Share


Multiplex Approach in Classification, Diagnosis, and Prognostication in Acute Myeloid Leukemia: An Experience from Tertiary Cancer Center in South India
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(03): 266-272
DOI: DOI: 10.4103/ijmpo.ijmpo_89_16
Abstract
Introduction: Acute myeloid leukemia (AML) is a heterogeneous group of disorders classified as per FAB subtypes and more recently by WHO by underlying genetic abnormalities. Aims and Objectives: This study aims to analyze the morphology, immunophenotype, cytogenetic and molecular abnormalities in around 200 patients of AML diagnosed over a period of 7 years at our institute and to determine relative frequency of various subtypes (based on FAB and WHO classification). An attempt to characterize the associations between hematological parameters, immunophenotype and these subtypes was also made. Materials and Methods: All cases diagnosed as AML on morphology, cytochemistry and/or immunophenotyping and tested for recurrent genetic abnormalities during period of Jan 2008-July 2014 were included in the study. Results: Age of presentation was younger in our AML patients as compared to western literature. Amongst FAB and WHO subtypes, M2 and t (15;17) PML-RARA were the most common groups respectively. As expected, CD33, CD13, were the most commonly expressed markers followed by HLA-DR, CD117, CD34 and CD14. Aberrant expression was seen in 62(41.6%) cases, most common was CD7 (15.4%), followed by CD56 (14.8%), CD19 (6.7%) and CD2 (4.7%). Significant associations between immunophenotypic markers and FAB subtypes as well as WHO subtypes were established. Conclusion: This is a hospital based study, giving a detailed account of frequencies of AML subtypes, hematological parameters and immunophenotypic markers in AML patients at our institute. Being a large and one of its kind study to establish significant associations between various haematological and immunophenotypic parameters with respective AML subtypes and genetic abnormalities, it might prove to be very useful in Indian setup where facilities for cytogenetic analysis are not available in many laboratories.
Publication History
Article published online:
04 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Introduction:
Acute myeloid leukemia (AML) is a heterogeneous group of disorders classified as per FAB subtypes and more recently by WHO by underlying genetic abnormalities.
Aims and Objectives:
This study aims to analyze the morphology, immunophenotype, cytogenetic and molecular abnormalities in around 200 patients of AML diagnosed over a period of 7 years at our institute and to determine relative frequency of various subtypes (based on FAB and WHO classification). An attempt to characterize the associations between hematological parameters, immunophenotype and these subtypes was also made.
Materials and Methods:
All cases diagnosed as AML on morphology, cytochemistry and/or immunophenotyping and tested for recurrent genetic abnormalities during period of Jan 2008-July 2014 were included in the study.
Results:
Age of presentation was younger in our AML patients as compared to western literature. Amongst FAB and WHO subtypes, M2 and t (15;17) PML-RARA were the most common groups respectively. As expected, CD33, CD13, were the most commonly expressed markers followed by HLA-DR, CD117, CD34 and CD14. Aberrant expression was seen in 62(41.6%) cases, most common was CD7 (15.4%), followed by CD56 (14.8%), CD19 (6.7%) and CD2 (4.7%). Significant associations between immunophenotypic markers and FAB subtypes as well as WHO subtypes were established.
Conclusion:
This is a hospital based study, giving a detailed account of frequencies of AML subtypes, hematological parameters and immunophenotypic markers in AML patients at our institute. Being a large and one of its kind study to establish significant associations between various haematological and immunophenotypic parameters with respective AML subtypes and genetic abnormalities, it might prove to be very useful in Indian setup where facilities for cytogenetic analysis are not available in many laboratories.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous group of disorders characterized by blocked or impaired differentiation of hematopoietic stem cells, resulting in abnormal accumulation of immature precursors. French–American–British (FAB) system has remained the reference method of classification for many years. WHO in 2008 incorporated genetic features into the classification of AML, thus making morphology, immunophenotyping, cytogenetic, and molecular techniques (MIC-M) the basis of AML subtyping. Approximately 60% of AML is now genetically classified.[1] Some of them are known to be closely associated with specific FAB subtypes. In this study, we have reported the frequencies of FAB and WHO subtypes at our institute. MIC-M features of AML were also analyzed to establish and confirm the association of FAB subtypes and their immunophenotypic features with underlying genetic abnormalities.
Aims and objectives
This study aims to determine the frequencies of FAB subtypes and genetic abnormalities among AML patients at our institute, to analyze the MIC-M features and hematological parameters of AML patients, and to characterize the association among them.
Materials and Methods
All cases diagnosed as AML on morphology, cytochemistry, and/or immunophenotyping and tested for recurrent genetic abnormalities during January 2008–July 2014 were included in the study. Cases diagnosed as AML on morphology, cytochemistry, and/or immunophenotyping but not worked up further for genetic abnormalities were excluded from the study.
Morphology and cytochemical stains
Peripheral blood (PB) and bone marrow (BM) smears were stained by Leishman and Giemsa stain, and cytochemistry was performed by standard methods (myeloperoxidase [MPO], nonspecific esterase [NSE]).
Immunophenotyping
Flow cytometry was performed on PB/BM on a four-color flow cytometer (FACS Calibur [Becton Dickinson, San Jose, CA, USA]) using standard protocol. Monoclonal antibodies used in this study included fluorescein isothiocyanate, phycoerythrin, or peridinin-chlorophyll-protein, allophycocyanin-labeled CD45, CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD13, CD14, CD19, CD22, CD33, CD34, CD38, CD56, CD117, human leukocyte antigen-antigen D-related (HLA-DR), and isotype control IgGs.
Cytogenetic studies and fluorescence in situ hybridization (FISH) were performed in AML cases; singly or both. FISH for AML1/ETO and PML/RARA was performed using Vysis LSI dual color, dual fusion translocation probes while Vysis LSI dual color, break apart probes were used for MLL and Inv 16 rearrangement as per manufacturer's instructions. Cytogenetic studies were performed by setting up unstimulated cultures of PB/BM, G-banded and analyzed according to the International System for Human Cytogenetic Nomenclature guidelines. At least 20 metaphases were analyzed.
Molecular tests
Polymerase chain reaction (PCR) for FMS-like tyrosine kinase 3 (FLT3), nucleophosmin 1 (NPM1), and CCAAT-enhancer binding protein alpha (CEBPA) mutations was carried out in the National Accreditation Board for Testing and Calibration Laboratories accredited laboratories. FLT3/internal tandem duplication (ITD) mutations were assessed using specific primers for exons 11, 12 and D835 mutation by restriction fragment length polymorphism-mediated assay using primers flanking mutation site. For NPM1 mutation, amplification of exon 12 of NPM1 gene was carried out followed by analysis of PCR products using automated DNA sequencing.
Statistical methods
The data were subjected to statistical analysis using unpaired Student's t-test and Chi-square test. Treatment and outcomes were not analyzed in this study.
Results
Patient data
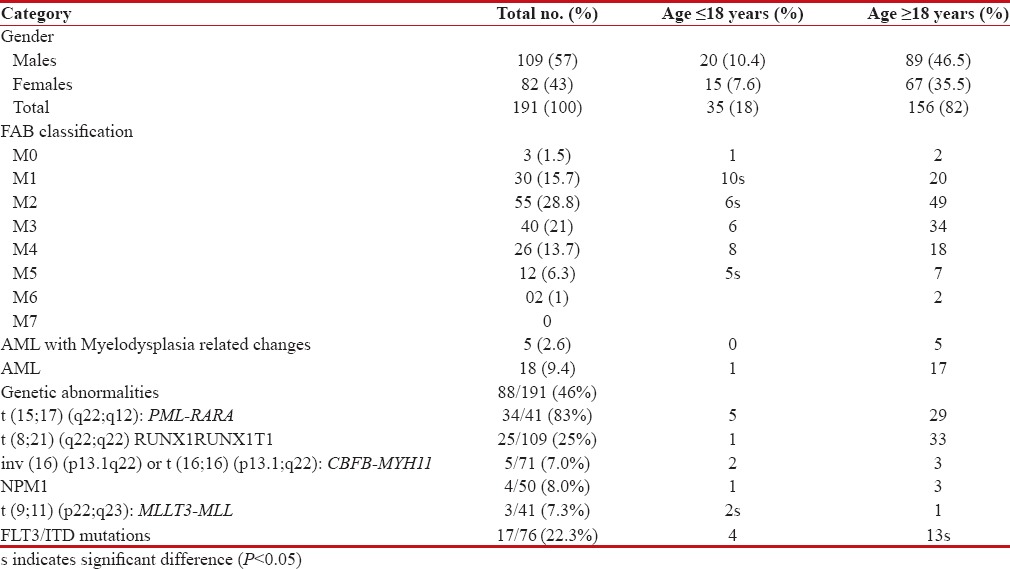
A total of 191 AML cases were tested for recurrent genetic abnormalities. The male-to-female ratio was 1.3:1 (109 males: 82 females). Age at diagnosis ranged from 2 to 74 years with a mean of 36.9 years. 156 (82%) patients were adults (18 years or older) while the remaining 35 (18%) patients were children (18 years or younger). Distribution of FAB and WHO subtypes among children and adults is shown in Table 1.
Table 1
Distribution of FAB and WHO subtypes amongst children and adults
|
Morphological analysis
The morphological subtypes in order of frequency among AML cases were M2 (55/191, 28.8%), M3 (40/191, 21%), M1 (30/191, 15.7%), M4 (26/191, 13.7%), M5 (12/191, 6.3%), M0 (3/191, 1.5%), and M6 (2/191, 1%). No M7 case was observed in the present study. There were 18 cases of AML, in which FAB subtyping could not be done. All the morphological subtypes were more common in adults than in children; a significant association was seen only in M1, M2, and M5 (P < 0>
Immunophenotypic analysis
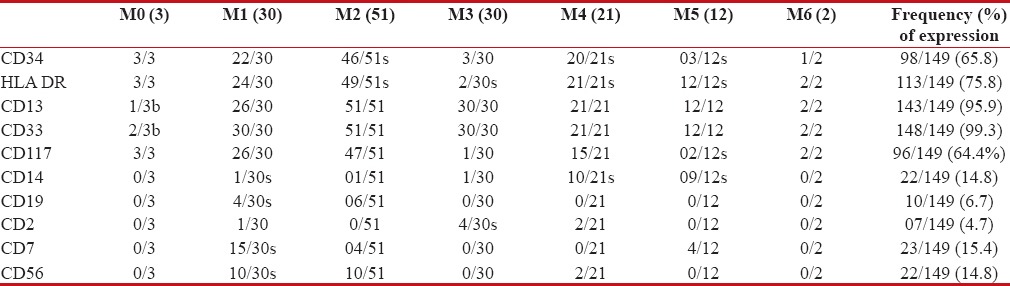
Immunophenotype was available in 149 cases. The 149 cases included 30 cases of M3, 3 cases of M0, 30 cases of M1, 51 cases of M2, 21 cases of M4, 12 cases of M5, and 2 cases of M6. Detailed immunophenotypic profile of FAB subtypes of AML cases is shown in Table 2.
Table 2
Immunophenotypic data of AML cases according to FAB subtypes
|
Precursor markers HLA-DR, CD117, and CD34 expressed at the overall rate of 75.8%, 64.4%, and 65.8%-respectively; however, in acute promyelocytic leukemia (APL) cases, CD34 and HLA-DR were expressed in 10%-and 6.6%-cases, respectively. As expected, CD33 and CD13 were expressed in highest percentage of cases, 99.3%-and 95.9%, respectively. CD 14 expression was observed in 14.8%-cases. Aberrant expression was seen in 62 (41.6%)-cases; most common was CD7 (15.4%), followed by CD56 (14.8%), CD19 (6.7%), and CD2 (4.7%). It was also observed that among FAB subtypes, CD56, CD7, and CD19 were most commonly expressed in M1 subtype (P < 0 xss=removed>P < 0>
Genetic abnormalities
Genetic abnormalities were detected in 46% (88 of 191) of AML cases. AML with t(15:17) PML/RARA (APL) formed the most common group. Thirty-four of 41 (83%) cases classified as APL were detected positive for t(15;17) [Figure 1]. Four cases were negative by FISH; however, they showed typical morphology of APL and tested positive for bcr1 isoform of PML/RARA by reverse transcription-PCR (RT-PCR).
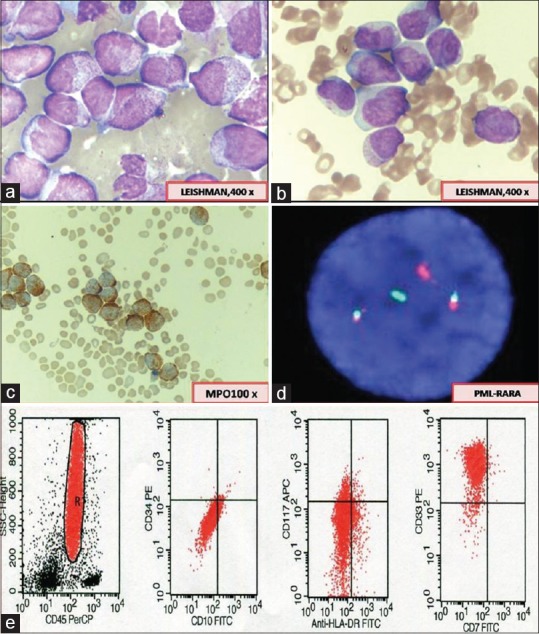
| Figure 1:(a) Abnormal promyelocytes (hypergranular acute promyelocytic leukemia) showing irregular nuclei and densely packed granules in the cytoplasm. (b) Hypogranular variant of acute promyelocytic leukemia showing apparent paucity of granules and predominantly bilobed nuclear shape. (c) Myeloperoxidase reaction showing strong positivity in leukemic promyelocytes. (d) Fluorescence in situ hybridization for t(15;17) promyelocytic leukemia/retinoic acid receptor alpha using Vysis LSI Dual Color, dual fusion translocation showing two fusion signals indicating presence of translocation. (e) Flow cytometry scatter plots showing characteristic lack of CD34 and human leukocyte antigen-antigen D related and homogenous bright expression of CD33 in acute promyelocytic leukemia cases
t(8;21)(q22;q22) was detected in 25% of the AML cases [Figure 2b]. As expected, all the cases belonged to M2 subtype, except for one case of M1.

| Figure 2:(a) A case of t(8;21) showing large blasts with abundant basophilic cytoplasm, some containing azurophilic granules and perinuclear hofs. Auer rods are seen in the cytoplasm as single long and sharp rod with tapered ends. (b) Fluorescence in situ hybridization for acute myeloid leukemia 1-ETO; t(8;21)(q22;q22) using Vysis LSI Dual Color, dual fusion translocation probes showing two fusion signals indicating presence of translocation. (c) Flow cytometry scatter plots in a case of t(8;21) showing high-intensity expression of CD34 along with CD19 and CD56 expression
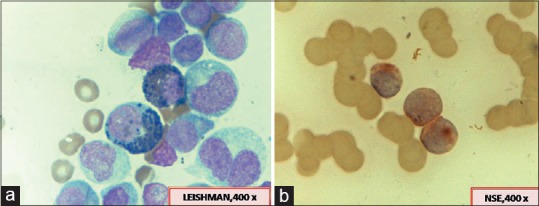
MLL and Inv 16 (core-binding factor beta [CBFB]-MYH11) were detected in 7.3% and 7%-cases, respectively [Figure 4a and
andb].b]. Distribution of FAB subtypes in these cases is as follows: Inv 16 (four cases M4, one case M5); MLL (two cases M4, one case M5).
| Figure 3:Fluorescence in situ hybridization for mixed lineage leukemia (a) and Inv 16 (b) using Vysis LSI dual color, break apart rearrangement probes showing the presence of respective gene rearrangements. (c) Flow cytometry scatter plots showing bright expression of CD14 indicating monocytic lineage of blasts in these cases
Characteristic morphological features, reactions with MPO/NSE, important immunophenotypic findings in our cases with t(15;17), t(8;21), and Inv16/MLL are shown in Figure 1a–e, Figure 2a–c, Figures Figures33 and and4,4, respectively.
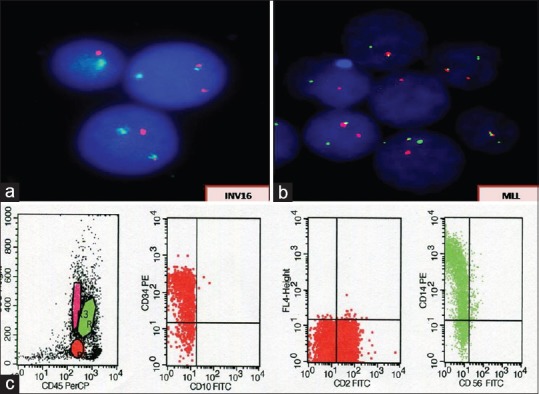
| Figure 4:(a) A case of acute myeloid leukemia with inv 16 showing the presence of abnormal eosinophils in addition to usual morphological features of myelomonocytic leukemia. (b) Monocytic nature of the blasts being highlighted by positive reaction with nonspecific esterase
Mutation analysis for FLT3 and NPM1 gene mutations was performed in 76, 50, and 34-cases, respectively. FLT3/ITD mutations were detected in 22.3%- cases which included seven patients of M2, five of M4, three of M1, and one each of M6 and AML-not otherwise specified. Frequency of FLT3/ITD in patients with normal karyotype (NK) was 21%, while none of the patients with abnormal karyotype showed this mutation, difference being highly significant (P < 0>
A total of 16 (94.1%) cases had FLT3Mt/NPM1wt profile, while 1 (5.9%) case showed FLT3Mt/NPM1Mt. Cases with FLT3/ITD mutation had strikingly male predominance and mostly found in adults, but there was no significant association (P > 0.05).
NPM1 mutations were detected in 8% (4/50) cases which included two cases of M2 and one case each of M1 and M6 subtype. FLT3/ITD mutations were present in 25%-cases of NPM1Mt and 21%-of NPM1wt phenotype, but difference was insignificant (P > 0.05).
Thirty-four patients were tested for CEBPA; however, none of the patients tested positive for this mutation.
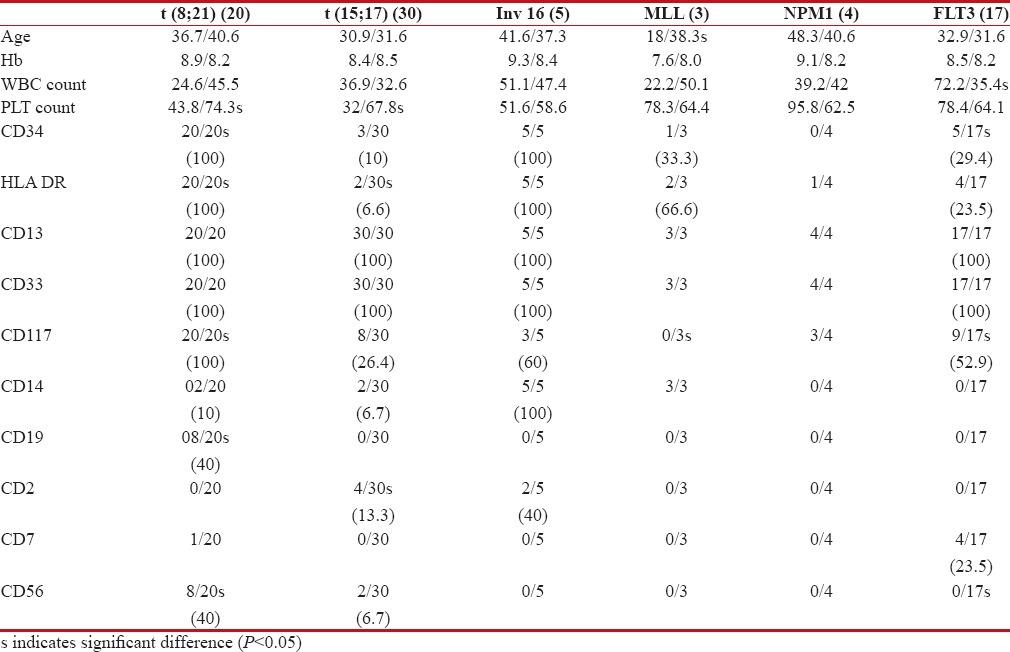
Data on age, hemoglobin (Hb), white blood cell (WBC) count, platelet count, and immunophenotype of respective group of genetic abnormality are shown in Table 3.
Table 3
Data on age, Hb, WBC count, platelet count, and immunophenotype of respective recurrent genetic abnormalities
|
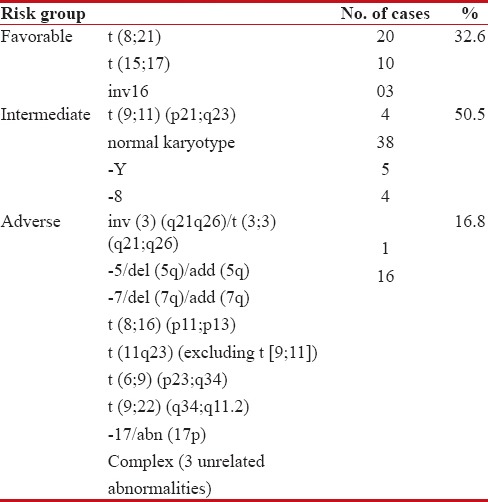
Cytogenetic analysis was done in all the 191 cases; informative karyotype (normal or abnormal) was available in 101 (52.8%) cases. Reasons for uninformative karyotypes could be insufficient sampling, no analyzable metaphases owing to previous treatment with antibiotics, blasts <10>Table 4.
Table 4
Risk stratification of AML cases based on cytogenetic profile
|
In NK patients, 8/38 (21%)-cases showed FLT3 mutations, 1/38 (2.6%)-case revealed NPM1 mutation, and 1 case each showed amplification of AML1, ETO, CBF, and MLL gene.
Some rare cytogenetic abnormalities were also observed in our study. These included t(11;18) and interstitial del 9q and near-tetraploidy as sole abnormalities in one case each.
Correlation of immunophenotype with French–American–British subtypes, recurrent genetic abnormalities, and hematological parameters
Among the FAB subtypes, CD19/CD56/CD7 expression was significantly associated with M1 subtype and M2 subtype was significantly associated with CD34, HLA-DR, and CD117 (P < 0 xss=removed>P > 0.05). Other markers found to be of significant associated were lack of CD34 and HLA-DR and expression of CD2 in M3 subtypes and CD14 in M4 and M5 subtypes.
Most of the cases of AML with t(8;21) belonged to M2 subtypes (96%) and were found to be significantly associated with low platelet count, expression of CD34, HLA-DR, CD117, CD19, and CD56. Cases with t(15;17) were significantly associated with low platelet count, absence of HLA-DR, and expression of CD2. Significant association was seen between younger age and presence of MLL translocations.
FLT3-ITD mutation was associated with significantly elevated WBC counts compared to wild-type (wt) FLT3 (P < 0>
Discussion
In the current study, mean age at presentation was 36.9 years (range 2–74 years) which is much younger than described in the literature (58–63 years).[2] CD 117 expression was seen in 64.4%-cases of AML similar to that reported in the literature; however, no correlation was apparent between CD117 expression and the degree of myeloid differentiation by FAB subtypes.[3] In this study, we found a significant association of CD117 expression with M1, M2 FAB subtypes and also with (8:21) translocation. One characteristic feature of t(8;21) AML is a striking myeloid immaturity, such as weak or absent expression of CD33[4] consistent with the suggestion that the AML1/ETO fusion event occurs at an early progenitor cell stage.[5] Further, CD13/CD33 expression was seen in 100%-cases which may be explained by majority of our cases being of M2 subtype.
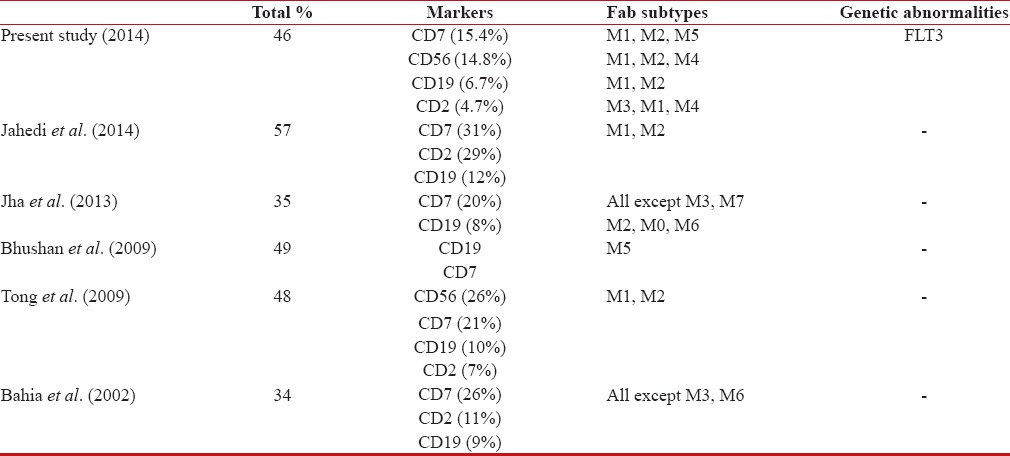
Aberrant expression of immunophenotypic markers was seen in 46%-cases in our study, CD7 being the most commonly expressed marker. Furthermore, expression of CD7 was found to be associated with FLT3 mutations. Table 5 shows a comparison with other studies with respect to aberrantly expressed markers.
Table 5
Aberrant phenotypic expression and comparison with other studies
|
AML with t(15;17) PML/RARA was significantly associated with lack of HLA-DR expression and presence of CD2 expression. CD117 expression in APL cases is an important question to be answered as it often is misinterpreted as negative because of weak intensity but forms an excellent tool for monitoring minimal residual disease in APL cases because of its invariable presence.[6] We found CD117 expression in 26.4% of APL cases, lower than reported in literature (50.7%).[7]
Genetic abnormalities were detected in 46% of the cases, comparable with that previously reported in the literature ranging from 52% to 80%.[4,5,8] t(15;17) PML/RARA formed the most common genetic abnormality in our study. Further, t(8;21) represented the next most frequent group seen in 25% of AML patients, followed by NPM1, MLL, and Inv 16 (CBFB-MYH11) detected in 8%, 7.3%, and 7%-cases, respectively. Overall frequency of t(15:17) and t(8;21) was found to be higher in our study as compared to that reported in the literature before. It can be attributed to selection bias based on morphological subtype M3 and M2, respectively. We found inv (16) in 7%-of patients, consistent with the results from previous studies (ranging from 2% to 8%).
Recent studies have demonstrated incidence of FLT3 mutation varying from 20% to 35% of all AML and up to 40%-of patients with NK in western countries.[9] In India, published data till date are sparse. Ahmad et al.[10] described incidence of FLT3 mutation to be 23.3%-in Indian population which is similar to that observed in our study (22.3%). FLT3/tyrosine kinase domain mutations were identified in 5.6%-cases in our study comparable to 8.8%-reported by another study from India.[10]
Mutation in the CEBPA gene has been reported as being one of the common genetic abnormalities in AML and is associated with a good clinical outcome. Ahmed et al.[11] reported CEBPA mutations in 8.3% of Indian AML patients; however, none of 34-patients tested for CEBPA positive in the current study.
In the current study, four cases of APL that were negative by FISH and cytogenetics however showed typical morphology and tested positive for PML/RARA fusion transcript by RT-PCR. The classic t(15;17) associated with PML/RARA fusion is detected in approximately 70–95%-of conventional cytogenetic studies of APL.[12] t(15;17) may occasionally not be observed in routine chromosome analyses in APL because of poor chromosome morphology, the presence of complex translocations, or submicroscopic insertions, leading to masked or cryptic PML/RARA fusions that are undetectable by metaphase analysis may give false negative results in these cases.[13] Therefore, it is critical that laboratories are aware of these rare cases of APL with false-negative FISH and cytogenetic results. Cases in which APL is suspected, but chromosomes and FISH results are normal, deserve further molecular analyses such as RT-PCR to provide definitive diagnoses.
AML represents a heterogeneous group of disorders. Frequency of different subtypes varies among different countries which can be explained by geographic and ethnic differences which play an important role in leukemogenesis of AML cases.
AML with t(8;21) displays an exclusive immunophenotyping with significantly high expression of CD19 and CD56 as well as precursor cell markers (CD34, CD117, and HLA-DR) and combination detection of CD34/HLA-DR/CD117/CD19/CD56 may be suggestive of t(8;21)(q22;q22) cytogenetic abnormality in Indian setup where facilities for cytogenetic analysis of such cases are not available.
The presence of CD2 expression should indicate APL especially hypogranular cases where the expected characteristic (high side scatter, high forward scatter) scattergram for M3 APL is not seen and should prompt for testing for PML/RARA fusion protein in otherwise doubtful cases.
Expression of CD7 along with CD117 may be taken as an indicator of FLT3 mutation which would be of great clinical impact; however, further studies on larger cohort of patients are suggested.
Finding of low frequency of CD34 is an important indicator of NPM1 mutant cases. Because of less number of cases in NPM1 group, no further conclusions could be drawn.
Cases suspected to be APL where chromosomes and FISH results are normal, deserve further molecular analyses such as RT-PCR to provide definitive diagnoses.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010;116:354-65.
- Cheng Y, Wang Y, Wang H, Chen Z, Lou J, Xu H, et al. Cytogenetic profile of de novo acute myeloid leukemia: A study based on 1432 patients in a single institution of China. Leukemia 2009;23:1801-6.
- Reuss-Borst MA, Bühring HJ, Schmidt H, Müller CA. AML: Immunophenotypic heterogeneity and prognostic significance of c-kit expression. Leukemia 1994;8:258-63.
- Andrieu V, Radford-Weiss I, Troussard X, Chane C, Valensi F, Guesnu M, et al. Molecular detection of t(8;21)/AML1-ETO in AML M1/M2: Correlation with cytogenetics, morphology and immunophenotype. Br J Haematol 1996;92:855-65.
- Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t (8;21) AML. Oncogene 2001;20:5660-79.
- Paietta E. Surrogate marker profiles for genetic lesions in acute leukemias. Best Pract Res Clin Haematol 2010;23:359-68.
- Liu Y, Chen YT, Li JX, Zhang SH. Immunophenotypic analysis of leukemia promyelocytes in 71 patients with acute promyelocytic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012;20:806-11.
- Heim S, Mitelman F. Cancer Cytogenetics: Chromosomal and Molecular Genetic Aberrations of Tumor Cells. 2nd ed. New York: Wiley-Liss; 1995. p. 72-3.
- Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002;100:59-66.
- Ahmad F, Mandava S, Das BR. Analysis of FLT3-ITD and FLT3-Asp835 mutations in de novo acute myeloid leukemia: Evaluation of incidence, distribution pattern, correlation with cytogenetics and characterization of internal tandem duplication from Indian population. Cancer Invest 2010;28:63-73.
- Ahmad F, Rajput S, Mandava S, Das BR. Molecular evaluation of CEBPA gene mutation in normal karyotype acute myeloid leukemia: A comparison of two methods and report of novel CEBPA mutations from Indian acute myeloid leukemia patients. Genet Test Mol Biomarkers 2012;16:707-15.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Geneva, Lyon: World Health Organization, IARC Press; 2008.
- Brockman SR, Paternoster SF, Ketterling RP, Dewald GW. New highly sensitive fluorescence in situ hybridization method to detect PML/RARA fusion in acute promyelocytic leukemia. Cancer Genet Cytogenet 2003;145:144-51.
| Figure 1:(a) Abnormal promyelocytes (hypergranular acute promyelocytic leukemia) showing irregular nuclei and densely packed granules in the cytoplasm. (b) Hypogranular variant of acute promyelocytic leukemia showing apparent paucity of granules and predominantly bilobed nuclear shape. (c) Myeloperoxidase reaction showing strong positivity in leukemic promyelocytes. (d) Fluorescence in situ hybridization for t(15;17) promyelocytic leukemia/retinoic acid receptor alpha using Vysis LSI Dual Color, dual fusion translocation showing two fusion signals indicating presence of translocation. (e) Flow cytometry scatter plots showing characteristic lack of CD34 and human leukocyte antigen-antigen D related and homogenous bright expression of CD33 in acute promyelocytic leukemia cases
| Figure 2:(a) A case of t(8;21) showing large blasts with abundant basophilic cytoplasm, some containing azurophilic granules and perinuclear hofs. Auer rods are seen in the cytoplasm as single long and sharp rod with tapered ends. (b) Fluorescence in situ hybridization for acute myeloid leukemia 1-ETO; t(8;21)(q22;q22) using Vysis LSI Dual Color, dual fusion translocation probes showing two fusion signals indicating presence of translocation. (c) Flow cytometry scatter plots in a case of t(8;21) showing high-intensity expression of CD34 along with CD19 and CD56 expression
| Figure 3:Fluorescence in situ hybridization for mixed lineage leukemia (a) and Inv 16 (b) using Vysis LSI dual color, break apart rearrangement probes showing the presence of respective gene rearrangements. (c) Flow cytometry scatter plots showing bright expression of CD14 indicating monocytic lineage of blasts in these cases
| Figure 4:(a) A case of acute myeloid leukemia with inv 16 showing the presence of abnormal eosinophils in addition to usual morphological features of myelomonocytic leukemia. (b) Monocytic nature of the blasts being highlighted by positive reaction with nonspecific esterase
References
- Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010;116:354-65.
- Cheng Y, Wang Y, Wang H, Chen Z, Lou J, Xu H, et al. Cytogenetic profile of de novo acute myeloid leukemia: A study based on 1432 patients in a single institution of China. Leukemia 2009;23:1801-6.
- Reuss-Borst MA, Bühring HJ, Schmidt H, Müller CA. AML: Immunophenotypic heterogeneity and prognostic significance of c-kit expression. Leukemia 1994;8:258-63.
- Andrieu V, Radford-Weiss I, Troussard X, Chane C, Valensi F, Guesnu M, et al. Molecular detection of t(8;21)/AML1-ETO in AML M1/M2: Correlation with cytogenetics, morphology and immunophenotype. Br J Haematol 1996;92:855-65.
- Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t (8;21) AML. Oncogene 2001;20:5660-79.
- Paietta E. Surrogate marker profiles for genetic lesions in acute leukemias. Best Pract Res Clin Haematol 2010;23:359-68.
- Liu Y, Chen YT, Li JX, Zhang SH. Immunophenotypic analysis of leukemia promyelocytes in 71 patients with acute promyelocytic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012;20:806-11.
- Heim S, Mitelman F. Cancer Cytogenetics: Chromosomal and Molecular Genetic Aberrations of Tumor Cells. 2nd ed. New York: Wiley-Liss; 1995. p. 72-3.
- Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002;100:59-66.
- Ahmad F, Mandava S, Das BR. Analysis of FLT3-ITD and FLT3-Asp835 mutations in de novo acute myeloid leukemia: Evaluation of incidence, distribution pattern, correlation with cytogenetics and characterization of internal tandem duplication from Indian population. Cancer Invest 2010;28:63-73.
- Ahmad F, Rajput S, Mandava S, Das BR. Molecular evaluation of CEBPA gene mutation in normal karyotype acute myeloid leukemia: A comparison of two methods and report of novel CEBPA mutations from Indian acute myeloid leukemia patients. Genet Test Mol Biomarkers 2012;16:707-15.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Geneva, Lyon: World Health Organization, IARC Press; 2008.
- Brockman SR, Paternoster SF, Ketterling RP, Dewald GW. New highly sensitive fluorescence in situ hybridization method to detect PML/RARA fusion in acute promyelocytic leukemia. Cancer Genet Cytogenet 2003;145:144-51.