 PDF
PDF  Views
Views  Share
Share


Metastatic Epithelioid Malignant Peripheral Nerve Sheath Tumor in a Known Case of Neurofibromatosis 1, Cytomorphological Appearance, and Critical Analysis of Immunohistochemistry
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(03): 387-390
DOI: DOI: 10.4103/ijmpo.ijmpo_113_17
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) are rare soft tissue tumors commonly arising from nerve roots in the extremities with a predilection in neurofibromatosis (NF) patients. MPNSTs are known to behave aggressively, with worse prognosis in nonresectable cases, and a 5-year survival of about 52% and 15% in resectable and unresectable cases, respectively. Although cytological diagnosis in a known case of NF-1 is possible, however, in the absence of any associated syndrome in a sporadic case, it is very challenging to accurately diagnose this tumor. Till date, to the best of our knowledge, only three cases of epithelioid MPNST correctly diagnosed on cytological examination have been described. We are presenting another case of epithelioid MPNST in a 40-year-old patient with stigmata of NF-1 since childhood, diagnosed on fine-needle aspiration cytology from the left flank mass with subsequent histopathology from left inguinal lymph node metastasis.
Keywords
Cytology - epithelioid - fine-needle aspiration cytology - malignant peripheral nerve sheath tumors - neurofibromatosis-1Publication History
Article published online:
04 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) are rare soft tissue tumors commonly arising from nerve roots in the extremities with a predilection in neurofibromatosis (NF) patients. MPNSTs are known to behave aggressively, with worse prognosis in nonresectable cases, and a 5-year survival of about 52% and 15% in resectable and unresectable cases, respectively. Although cytological diagnosis in a known case of NF-1 is possible, however, in the absence of any associated syndrome in a sporadic case, it is very challenging to accurately diagnose this tumor. Till date, to the best of our knowledge, only three cases of epithelioid MPNST correctly diagnosed on cytological examination have been described. We are presenting another case of epithelioid MPNST in a 40-year-old patient with stigmata of NF-1 since childhood, diagnosed on fine-needle aspiration cytology from the left flank mass with subsequent histopathology from left inguinal lymph node metastasis.
Introduction
Malignant peripheral nerve sheath tumors (MPNST) as their name suggests are derived from nerve sheath cells either Schwann cell or perineurial cells. They are also known as neurofibrosarcoma, neurogenic sarcoma, and malignant schwannoma and account for 5% of malignant soft tissue tumors. MPNST is a very aggressive tumor usually arises from nerve sheath of peripheral nerve roots, and commonly affects extremities. These tumors also show frequent association with neurofibromatosis-1 (NF-1)[1,2] and occur along with its benign counterpart neurofibroma or arise from it. Since MPNSTs recapitulate appearance of various cells of nerve sheath, its low power histomorphology may vary from neurofibroma to fibrosarcoma. Rhabdomyoblastic, glandular, and epithelial differentiation has been also described in rare cases of MPNSTs.[3] Although few case series describing histological features of epithelioid MPNST are reported, cytological characteristics and histocytological correlation are described only in three reported cases.[4,5] We report a case of epithelioid MPNST in a background of NF-1 with natural course of its progression diagnosed on fine-needle aspiration cytology (FNAC) with histocytological correlation and immunohistochemical characterization.
Case Report
A 40-year-old male patient presented to the Outpatient Department of Surgery with the complaints of swelling in the left flank for 20 years. The patient was apparently well 20 years back when he noticed the swelling which was nontender, gradual in onset, and progressively increasing in size. The patient operated in a local hospital; however, documentation and pathological details of the surgical interventions were not available. In the last 1½ month period, he redeveloped a swelling in the same region and presented in our hospital. On examination, the patient was found to have multiple variable-sized neurofibromas throughout the body along with few café au lait spots. Except anorexia, no history of fever/vomiting/neurological symptoms/weight loss or clubbing was present. The systemic examination revealed bilateral decreased air entry with left zonal crepts. A large hard, nontender, nonpulsatile, nonreducible, nonfluctuant, fixed lump measuring 17 cm × 15 cm was identified in the left lumbar region [Figure 1]. In addition, multiple axillary and inguinal lymph nodes also noted largest measuring approximately 2 cm in diameter. The family history was noncontributory. Hemogram, liver function test, and kidney function test were within normal limits.
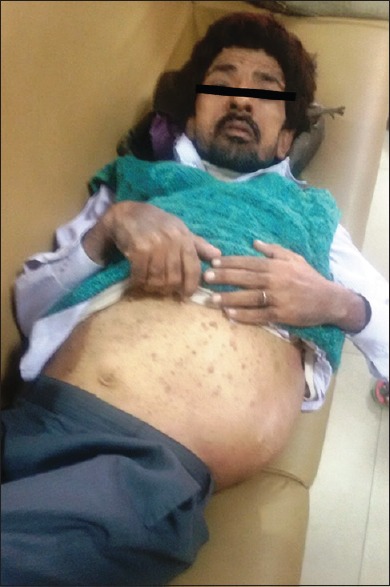
| Figure 1:Clinical image of the patient showing multiple neurofibromas all over the body with a large mass in the left flank
Magnetic resonance imaging (MRI) showed a well-defined round to oval large mass measures 9.7 cm × 7.5 cm in deep subcutaneous and intermuscular compartment of the left posterolateral abdominal wall. The tumor was heterogeneously hyperintense on T1 weighted and T2 weighted as compared to muscles [Figures [Figures11 and and2a2a–d].

| Figure 2:Magnetic resonance imaging T1-weighted coronal (a), T2-weighted coronal (b), T2-weighted axial (c), and STIR axial (d) images showing a well-defined round to oval large mass (arrow) in deep subcutaneous and intermuscular compartment of left posterolateral abdominal wall. The lesion measures 9.7 cm × 7.5 cm in transverse and anteroposterior dimension, respectively. The lesion is heterogeneously hyperintense on T1 weighted and T2 weighted as compared to muscles. There is no signal drop on fat suppressed sequence suggestive of absent fatty component. The central part of lesion shows T2 hyperintense areas suggestive of necrosis. There is no extension into spinal canal. Chest X-ray posteroanterior view, (e) left-sided pleural effusion
FNAC smears were cellular and showed a tumor comprising of dispersed large atypical cells with eccentric nucleus, marked nuclear pleomorphism, binucleation, multinucleation, nuclear lobulations/folding, occasional nuclear pseudoinclusions, coarsely granular chromatin, and abundant amount of basophilic cytoplasm [Figure 3]. In addition, many mitotic figures and few degenerated cells were also noted.
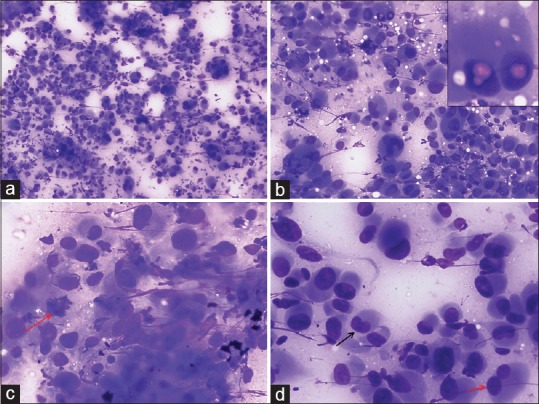
| Figure 3:Fine-needle aspiration cytology images showing (a) A cellular tumor comprising of singly dispersed pleomorphic epithelioid cells (Giemsa, ×100), (b) Tumor cells showing marked variation in cell size with numerous binucleated and multinucleated cells. Occasional intranuclear pseudoinclusions (inset) and cytoplasmic vacuolization are present (Giemsa, ×200), (c) Tumor cells displaying eccentric nucleus, coarse granular chromatin, and abundant basophilic cytoplasm. Arrow indicates a mitotic figure (Giemsa, ×400), (d) Image displaying variation in nuclear shape and size with arrows indicating a reniform nucleus (black) and a bilobed nucleus (red) (Giemsa, ×400)
Chest X-ray showed left-sided pleural effusion [Figure 2e], and fluid cytology was reported outside as malignant, likely sarcoma. An excision biopsy was done from one of the inguinal lymph nodes. The histological section showed complete effacement of the nodal architecture and infiltration by a tumor arranged in sheets with few remnants lymphoid follicles [Figure 4a]. The cells showed eccentrically placed large nucleus with vesicular chromatin, prominent nucleoli, and abundant amount of eosinophilic cytoplasm. Frequent binucleation/multinucleation, nuclear lobulations, mitotic figures, and perinodal spread were noted [Figure 4b–d]. The tumor cells revealed diffuse immunopositivity for S-100 [Figure 4e] and vimentin [Figure 4f] (Both Thermo-Scientific, RTU), however negative for cytokeratin, epithelial membrane antigen, CD20, CD30, CD68, desmin, CD34, smooth muscle actin (All Thermo-Scientific, RTU), HMB-45, Melan-A, and CD3 (All Dako, RTU). INI-1 (Cell Marque; dilution 1:100) immunohistochemistry (IHC) did not show loss of expression in the tumor cells. Based on characteristic cytology, histomorphology, and IHC in a background of NF-1, a final diagnosis of epithelioid variant of MPNST with preserved INI-1 status was made. Unfortunately, the patient did not receive any chemotherapy or radiation therapy and succumbed after 1 month.
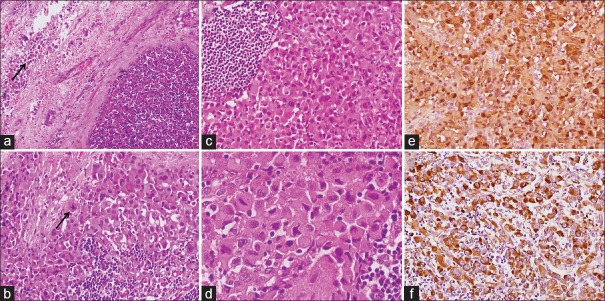
| Figure 4:Histomorphological and immunohistochemical images from inguinal lymph node metastases showing (a) Effacement of the nodal architecture with perinodal spread (arrow) (H and E, ×100), (b) Tumor cells arranged in diffuse sheets admixed with lymphocytes and a mitotic figure (arrow) (H and E, ×200), (c) A remnant lymphoid follicle in the left (H and E, ×200), (d) High-power image showing marked cellular pleomorphism, eccentrically lying nucleus, coarse granular chromatin, and abundant amount of cytoplasm (H and E, ×400), (e) and (f) Diffuse S-100 and vimentin positivity in tumor cells, respectively (immunohistochemistry, ×200)
Discussion
Epithelioid variant of MPNST is a very rare tumor which accounts for 5% of all the malignant neural tumors.[4] The incidence of MPNSTs is almost equal between NF1 patients and sporadic cases. Radiation exposure has also been described as a cause of MPNSTs.[6] Few series describing epithelioid variant of MPNSTs are published in the English literature.[7,8] However, cytomorphological studies are very scant, and except for a single case series,[9] only few case reports are available. Moreover, the rarity of epithelioid variant further limits description of its cytological counterpart; at present, only three cases in two reports have presented FNAC details.[4,5] Klijanienko et al. in their series also included one case of epithelioid MPNST, but initially, on cytology, it was reported as sarcoma, not otherwise specified which later confirmed on histology.[9] Like its conventional histological (spindle cell) counterpart, epithelioid MPNST usually arises from major nerve roots and commonly located in trunk, extremities, and limb girdles. They may arise from or associated with a preexisting benign nerve sheath tumor such as neurofibroma. In this case, whether tumor aroused de novo or developed in a preexisting neurofibroma was uncertain because of unavailability of pathological details of previous surgeries. The tumor developed over a long period of 20 years with recurrence and later distant metastases into inguinal and axillary lymph nodes and malignant pleural effusion. The most distinct feature of these tumors is their cytomorphological characteristics which distinguish them from the both benign and malignant conventional counterparts. However, due to the similarities of cytological appearance with melanomas, epithelioid sarcoma, poorly differentiated carcinoma, and other epithelioid soft tissue sarcomas, they should be carefully diagnosed considering clinical, radiological, and immunohistochemical aspects. Since available literature is very limited, no defined cytological criteria have been established. Moreover, the cytological mimicry of these tumors with many other relatively more common tumors is the challenging issue for defining the FNAC characteristics. Dodd et al.[4] found remarkable cytological differences between their two cases. Both of their cases were sparsely cellular; however, one comprise of mainly round singly dispersed cells without any stroma and other showed loosely coherent cells in clumps with fragments of myxoid stroma, and cells were more obviously plump spindled rather than round. Jiwani et al.[5] found richly cellular tumor which consists of pleomorphic cells with both spindle and epithelioid morphology showing prominent nucleoli and many mitotic figures with scant pink mesenchymal matrix material in the background. In our case, the aspirate was predominantly hemorrhagic with good cellularity in only one of the smear. The tumor comprising of dispersed singly lying as well as loosely cohesive clusters of markedly pleomorphic cells with epithelioid morphology. No spindle cell component or background stromal material was found. The cells delineated eccentrically placed nucleus with frequent binucleation/multinucleation, reniform nuclei, nuclear lobulations, coarsely granular chromatin, inconspicuous nucleoli, and abundant amount of basophilic cytoplasm. The cell block was not prepared due to limitations of the material; thus, immunocytochemistry was not performed. The above discussed four cases themselves displayed a varying spectrum of cytomorphological features. Although in our case, the patient had definite stigmata of NF-1, the previously reported three cases occurred sporadically. Sparse cellularity, hemorrhagic aspirate, unavailability of definite diagnostic criteria, and simulation with numerous other tumors make cell block and immunocytochemistry imperative for accurate diagnosis. Furthermore, cytohistological correlation plays an important role for further confirmation of the diagnosis. About 50%–70% of these tumors show loss of INI-1 on IHC.[5] However, in this case, INI-1 expression was retained.
MPNSTs are known to behave aggressively, with worse prognosis in nonresectable cases. Most MPNSTs are high-grade sarcomas, with a high tendency of producing local recurrence and distant metastasis. Tumor size >5 cm, high mitotic count, previous history of NF-1, and incomplete resection are the major determinants of poor outcome.[1] Laskin et al.[8] found a relatively better outcome in superficially located tumors in comparison to deep-seated cases. Although earlier literatures described a more aggressive course in the MPNSTs which occurred in association with NF-1, recent studies showed no differences in progression in the comparable size tumors from NF patients and sporadic cases.[10,11]
Conclusion
FNAC is a simple, outpatient department-based procedure which can greatly help in definitive diagnosis of epithelioid variant of MPNST in spite of cytologic variability and unavailable defined cytological criteria. Ancillary studies (immunocytochemistry), if feasible, can further strengthen and ensure diagnosis. In cases with inadequate aspirate material, a cytohistological correlation may be required for confirmation of diagnosis.
Limitations
The exact nature of tumor whether aroused de novo or developed in a preexisting neurofibroma was not determined due to unavailability of previous pathological details. Furthermore, due to limited aspirate material, immunocytochemistry was not done; however, IHC was performed in metastatic lesion in the inguinal nodes.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to acknowledge Mr. Shahajad Ali (JRF) for collection of patient's details
References
- Weiss SW, Goldblum JR, Enzinger FM, editors. Enzinger and Weiss's Soft Tissue Tumors. St. Louis: Mosby Inc.; 2008.
- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986;57:2006-21.
- Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012;123:295-319.
- Dodd LG, Scully S, Layfield LJ. Fine-needle aspiration of epithelioid malignant peripheral nerve sheath tumor (epithelioid malignant schwannoma). Diagn Cytopathol 1997;17:200-4.
- Jiwani S, Gokden M, Lindberg M, Ali S, Jeffus S. Fine-needle aspiration cytology of epithelioid malignant peripheral nerve sheath tumor: A case report and review of the literature. Diagn Cytopathol 2016;44:226-31.
- Kourea HP, Bilsky MH, Leung DH, Lewis JJ, Woodruff JM. Subdiaphragmatic and intrathoracic paraspinal malignant peripheral nerve sheath tumors: A clinicopathologic study of 25 patients and 26 tumors. Cancer 1998;82:2191-203.
- Lodding P, Kindblom LG, Angervall L. Epithelioid malignant schwannoma. A study of 14 cases. Virchows Arch A Pathol Anat Histopathol 1986;409:433-51.
- Laskin WB, Weiss SW, Bratthauer GL. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am J Surg Pathol 1991;15:1136-45.
- Klijanienko J, Caillaud JM, Lagacé R, Vielh P. Cytohistologic correlations of 24 malignant peripheral nerve sheath tumor (MPNST) in 17 patients: The institut curie experience. Diagn Cytopathol 2002;27:103-8.
- ;Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant peripheral nerve sheath tumor: Analysis of treatment outcome. Int J Radiat Oncol Biol Phys 1998;42:351-60.
- Stark AM, Buhl R, Hugo HH, Mehdorn HM. Malignant peripheral nerve sheath tumours – Report of 8 cases and review of the literature. Acta Neurochir (Wien) 2001;143:357-63.
| Figure 1:Clinical image of the patient showing multiple neurofibromas all over the body with a large mass in the left flank
| Figure 2:Magnetic resonance imaging T1-weighted coronal (a), T2-weighted coronal (b), T2-weighted axial (c), and STIR axial (d) images showing a well-defined round to oval large mass (arrow) in deep subcutaneous and intermuscular compartment of left posterolateral abdominal wall. The lesion measures 9.7 cm × 7.5 cm in transverse and anteroposterior dimension, respectively. The lesion is heterogeneously hyperintense on T1 weighted and T2 weighted as compared to muscles. There is no signal drop on fat suppressed sequence suggestive of absent fatty component. The central part of lesion shows T2 hyperintense areas suggestive of necrosis. There is no extension into spinal canal. Chest X-ray posteroanterior view, (e) left-sided pleural effusion
| Figure 3:Fine-needle aspiration cytology images showing (a) A cellular tumor comprising of singly dispersed pleomorphic epithelioid cells (Giemsa, ×100), (b) Tumor cells showing marked variation in cell size with numerous binucleated and multinucleated cells. Occasional intranuclear pseudoinclusions (inset) and cytoplasmic vacuolization are present (Giemsa, ×200), (c) Tumor cells displaying eccentric nucleus, coarse granular chromatin, and abundant basophilic cytoplasm. Arrow indicates a mitotic figure (Giemsa, ×400), (d) Image displaying variation in nuclear shape and size with arrows indicating a reniform nucleus (black) and a bilobed nucleus (red) (Giemsa, ×400)
| Figure 4:Histomorphological and immunohistochemical images from inguinal lymph node metastases showing (a) Effacement of the nodal architecture with perinodal spread (arrow) (H and E, ×100), (b) Tumor cells arranged in diffuse sheets admixed with lymphocytes and a mitotic figure (arrow) (H and E, ×200), (c) A remnant lymphoid follicle in the left (H and E, ×200), (d) High-power image showing marked cellular pleomorphism, eccentrically lying nucleus, coarse granular chromatin, and abundant amount of cytoplasm (H and E, ×400), (e) and (f) Diffuse S-100 and vimentin positivity in tumor cells, respectively (immunohistochemistry, ×200)
References
- Weiss SW, Goldblum JR, Enzinger FM, editors. Enzinger and Weiss's Soft Tissue Tumors. St. Louis: Mosby Inc.; 2008.
- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986;57:2006-21.
- Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012;123:295-319.
- Dodd LG, Scully S, Layfield LJ. Fine-needle aspiration of epithelioid malignant peripheral nerve sheath tumor (epithelioid malignant schwannoma). Diagn Cytopathol 1997;17:200-4.
- Jiwani S, Gokden M, Lindberg M, Ali S, Jeffus S. Fine-needle aspiration cytology of epithelioid malignant peripheral nerve sheath tumor: A case report and review of the literature. Diagn Cytopathol 2016;44:226-31.
- Kourea HP, Bilsky MH, Leung DH, Lewis JJ, Woodruff JM. Subdiaphragmatic and intrathoracic paraspinal malignant peripheral nerve sheath tumors: A clinicopathologic study of 25 patients and 26 tumors. Cancer 1998;82:2191-203.
- Lodding P, Kindblom LG, Angervall L. Epithelioid malignant schwannoma. A study of 14 cases. Virchows Arch A Pathol Anat Histopathol 1986;409:433-51.
- Laskin WB, Weiss SW, Bratthauer GL. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am J Surg Pathol 1991;15:1136-45.
- Klijanienko J, Caillaud JM, Lagacé R, Vielh P. Cytohistologic correlations of 24 malignant peripheral nerve sheath tumor (MPNST) in 17 patients: The institut curie experience. Diagn Cytopathol 2002;27:103-8.
- ;Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant peripheral nerve sheath tumor: Analysis of treatment outcome. Int J Radiat Oncol Biol Phys 1998;42:351-60.
- Stark AM, Buhl R, Hugo HH, Mehdorn HM. Malignant peripheral nerve sheath tumours – Report of 8 cases and review of the literature. Acta Neurochir (Wien) 2001;143:357-63.