 PDF
PDF  Views
Views  Share
Share


Laboratory Workup of Hypereosinophilia
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(06): 602-610
DOI: DOI: 10.1055/s-0043-1761261
Abstract
Hypereosinophilia (HE) can be caused by a wide variety of non-hematologic (secondary or reactive) and hematologic (primary, clonal) disorders. Diagnosing hypereosinophilia/hypereosinophilic syndrome (HE/HES) is challenging due to the complex nature of disease manifestations and numerous underlying etiologies. Knowing that only rare cases are clonal, it is wise to rule out reactive conditions and proceed with molecular and other advanced tools. The exclusion of secondary causes needs a detailed clinical evaluation followed by a wide range of serological and imaging investigations. Once reactive eosinophilia has been ruled out, the diagnosis of primary HE/HES is made using a combination of morphologic examination of the blood and bone marrow, conventional cytogenetics, fluorescent in situ hybridization, flow-cytometry, and T-cell clonality evaluation to look for histopathologic or clonal evidence of an underlying hematological disorder. The accurate diagnosis of clonal eosinophilia-causing myeloid and lymphoid neoplasms and the identification of numerous gene rearrangements significantly enhance patient outcomes, because a proportion of these patients (such as PDGFRA and PDGFRB rearrangements) responds well to tyrosine kinase inhibitors. Considering the complex etiopathologies, the cost of testing, and the time involved, the workup needs to be tailored according to the urgency of the situation and the resources available. In urgent situations with organ damage, it is crucial to initiate appropriate management without waiting for the results of investigations. In contrast, in a resource-limited situation, it is acceptable to employ step-by-step rather than comprehensive testing to rule out the most common causes first. Here, we discuss various laboratory investigations employed in diagnosing HE/HES, highlighting their importance in different situations.
Keywords
hypereosinophilia - hypereosinophilic syndrome - mastocytosis - tyrosine kinase domain fusions - myelodysplastic syndromes - myeloproliferative neoplasmsAuthors' Contributions
D.S. wrote initial manuscript; S.S. revised and approved the final manuscript.
Publication History
Article published online:
17 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Hypereosinophilia (HE) can be caused by a wide variety of non-hematologic (secondary or reactive) and hematologic (primary, clonal) disorders. Diagnosing hypereosinophilia/hypereosinophilic syndrome (HE/HES) is challenging due to the complex nature of disease manifestations and numerous underlying etiologies. Knowing that only rare cases are clonal, it is wise to rule out reactive conditions and proceed with molecular and other advanced tools. The exclusion of secondary causes needs a detailed clinical evaluation followed by a wide range of serological and imaging investigations. Once reactive eosinophilia has been ruled out, the diagnosis of primary HE/HES is made using a combination of morphologic examination of the blood and bone marrow, conventional cytogenetics, fluorescent in situ hybridization, flow-cytometry, and T-cell clonality evaluation to look for histopathologic or clonal evidence of an underlying hematological disorder. The accurate diagnosis of clonal eosinophilia-causing myeloid and lymphoid neoplasms and the identification of numerous gene rearrangements significantly enhance patient outcomes, because a proportion of these patients (such as PDGFRA and PDGFRB rearrangements) responds well to tyrosine kinase inhibitors. Considering the complex etiopathologies, the cost of testing, and the time involved, the workup needs to be tailored according to the urgency of the situation and the resources available. In urgent situations with organ damage, it is crucial to initiate appropriate management without waiting for the results of investigations. In contrast, in a resource-limited situation, it is acceptable to employ step-by-step rather than comprehensive testing to rule out the most common causes first. Here, we discuss various laboratory investigations employed in diagnosing HE/HES, highlighting their importance in different situations.
Keywords
hypereosinophilia - hypereosinophilic syndrome - mastocytosis - tyrosine kinase domain fusions - myelodysplastic syndromes - myeloproliferative neoplasmsIntroduction
Peripheral blood and tissue eosinophilia can be caused by a heterogeneous group of hematological and non-hematological conditions. The normal peripheral blood eosinophil ranges from 1 to 5%, with absolute eosinophil count (AEC) less than 0.5 × 109/L, and the bone marrow eosinophil percentage ranges between 1 and 6%.[1]
Eosinophilopoiesis and Biology of Eosinophils
Eosinophils are derived from CD34+ multipotent hematopoietic stem cells that are driven by variable levels of transcription factors like GATA binding protein 1 (GATA-1), Ets (erythroblast transformation specific) factor- PU.1, friend of GATA protein 1 (FOG-1), and CCAAT/enhancer binding proteins (C/EBP family)——C/EBPα and C/EBPε. The amalgamation of these transcription factors and their malleable expression ultimately decides the lineage differentiation of stem cells. The combination of GATA-1 expression, low levels of Ets factor PU.1, FOG-1 downregulation, and alternate C/EBP family expression favor the stem cells to differentiate into eosinophil/mast cells progenitors (EoMP) over the others.[2] The interplay of cytokines and chemokines mediates eosinophils' further differentiation, migration, and activation. The EoMPs that contain interleukin-5 receptors (IL-5R) exclusively transform into special eosinophil-committed progenitors (EoPs). These EoPs are further activated by lineage-specific cytokine IL-5 that is the crucial amplifier of proliferation and terminal differentiation of eosinophils. The EoMPs that lack sufficient IL-5R develop into basophil progenitors (BaP) and mast cells. IL-5 is the cardinal cytokine synthesized by mast cells, activated Th2 lymphocytes, macrophages, natural killer cells, endothelial cells, epithelial cells, and fibroblast cells of both the innate and adaptive immune systems. Apart from the IL-5, other cytokines like IL-3, IL-4, granulocyte-macrophage colony-stimulating factor, etc. also contribute in this process ([Fig. 1]).
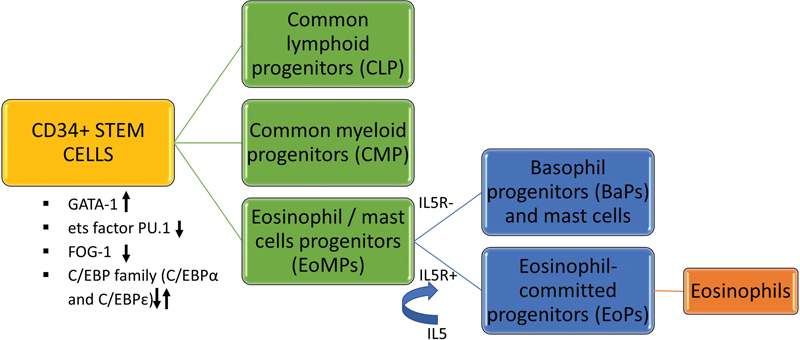
| Fig 1 : The process of eosinophilopoiesis.
Eosinophilia, Hypereosinophilia, Tissue Eosinophilia, and Hypereosinophilic Syndrome
Eosinophilia is defined as blood AEC >0.5 × 109/L. Based on the severity, they are graded as[1] mild (0.5–1.5 × 109/L), moderate (1.5–5.0 × 109/L), and severe (>5.0 × 109/L). Based on the duration of symptoms, they can be categorized into transient, episodic, or persistent. The term persistent pertains to peripheral blood eosinophilia that is observed or documented on at least two occasions at a minimum of 4 weeks interval. End-organ damage occurs more in persistent eosinophilia due to the protracted release of chemical mediators and their adverse biological effects. However, it does not mean that one can ignore transient and episodic eosinophilia, as they can also be life-threatening and warrant urgent lifesaving management.[1] Mild eosinophilia is seen in 3 to 10%-of the population, and the commonest causes are allergic diseases, drugs, and parasitic infestations. Hypereosinophilia (HE) is defined as persistent moderate-to-severe peripheral blood eosinophilia (>1.5 × 109/L) and/or tissue eosinophilia.[3]
Eosinophils are usually not seen in healthy tissues except for their presence in mucosa of the stomach, small and large bowels, uterus, thymus, spleen, and lymph nodes.[4] The criteria for tissue eosinophilia include bone marrow biopsy showing more than 20%-eosinophils of all nucleated cells and/or significant amount of tissue infiltration by eosinophils reported by expert pathologist and/or distinct eosinophil granule protein deposition in the tissue with or without extensive tissue infiltration by eosinophils.[3] HE can be diagnosed by hemogram findings and confirmed by peripheral blood film (PBF) examination. Patients with HE and organ dysfunction attributable to HE are diagnosed with hypereosinophilic syndrome (HES). The older criteria of HES by Simon et al[5] date back to 1975 that include blood AEC >1.5 × 109/L lasting more than 6 months, no identifiable secondary (reactive) causes of eosinophilia after complete investigations, and evidence of end-organ damage by persistent eosinophilia. The refined definition of HES is based on the changes made in the duration of the first criterion, that, blood AEC >1.5 × 109/L lasting more than 1 month.[3] HES with life-threatening organ dysfunction need immediate evaluation and intervention irrespective of the duration of HE.
Classification of HE and HES
They can be categorized into reactive or secondary HE/HES (common), primary or clonal HE/HES (rare), idiopathic HE/HES, and familial HE (very rare).[6] In both reactive and clonal eosinophilia, the key mediator for uncontrolled proliferation of eosinophils is IL-5, however, with different pathogenesis. The stimuli for the increased production of IL-5 in secondary eosinophilia are infections, drugs, allergies, and various malignancies (paraneoplastic or cytokine induces HE). The eosinophils are not clonal in this condition, though clonal cells like T cells can induce secondary eosinophilia. The most common cause of HE is reactive and allergic disorders. Reactive eosinophilia is predominantly mediated by IL-5 (along with IL-3 and IL-4) and is caused by atopic dermatitis, asthma and seasonal allergic disorders (rhinitis/hay fever), dermatological disorders like Wells syndrome (nonallergic granulomatous dermatitis), drug-induced (includes drug reaction with eosinophilia and systemic symptoms/DRESS), helminthic and fungal infections, primary gastrointestinal eosinophilic disorders, connective tissue disorders, rheumatological diseases, atheroembolic disease, Gleich syndrome, lymphoproliferative disorders, solid malignancies, and the lymphocytic variant of HE. The major secondary causes[7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] are summarized in [Table 1].
|
Atopy |
Asthma, allergic rhinitis, eczema |
|---|---|
|
Fungal infection |
Cryptococcus neoformans, Aspergillus spp, Coccidioides immitis, Paracoccidioides brasiliensis and Histoplasma capsulatum. |
|
Parasitic infection |
Anisakis simplex, Taenia solium, Schistosoma mansoni, Strongyloides stercoralis, toxoplasma,[9] Strongyloidiasis (Strongyloides stercoralis), Hookworm (Ancylostoma duodenale and Necator americanus), Filariasis (Loa loa, Wuchereria bancrofti, Mansonella perstans, Brugia malayi, onchocerca spp.), Ascariasis (Ascaris lumbricoides), Toxocariasis (Toxocara canis), Trichinosis (Trichinella spp.), Scabies (Sarcoptes scabiei), Fascioliasis (Fasciola hepatica) |
|
Viral |
HIV |
|
Immunological disorders |
Hyper-IgE syndrome, DOCK8 deficiency, PGM3 deficiency, STAT3 deficiency, CD40 deficiency, ADA deficiency, ZAP70 deficiency, CD3γ deficiency, MHC II deficiency, TCR-α deficiency, MALT1 deficiency, Kostmann disease, cyclic neutropenia, Omenn syndrome, Wiskott-Aldrich syndrome, autoimmune lymphoproliferative syndrome, immunodysregulation polyendocrinopathy enteropathy X-linked, Papillon-Lefevre syndrome, and CVID[10] |
|
Dermatological disorders |
Chronic spontaneous urticaria, atopic dermatitis, eosinophilic dermatoses, eosinophilic cellulitis (Wells syndrome), eosinophilic pustular folliculitis, eosinophilic fasciitis (Shulman disease), granuloma faciale; recurrent cutaneous eosinophilic vasculitis (RCEV)[11] |
|
Pulmonary diseases |
Idiopathic acute or chronic eosinophilic pneumonia, Loffler syndrome, allergic bronchopulmonary aspergillosis (ABPA), sarcoidosis |
|
Drugs |
NSAIDs, anticonvulsant and DRESS syndrome. Drugs implicated in DRESS syndromes are aromatic antiepileptic drugs like phenytoin, lamotrigine and carbamazepine, allopurinol, antimicrobial sulfonamides (sulfasalazine), and dapsone, other antibiotics such as vancomycin and minocycline[12] [13] [14] |
|
Connective tissue and rheumatological disorders |
Churg-Strauss syndrome, Wegener's granulomatosis, systemic lupus erythematosus, polyarteritis nodosa (PAN), eosinophilic fasciitis, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), dermatomyositis and Sjogren syndrome |
|
Allergic gastroenteritis and esophagitis, inflammatory bowel disease (ulcerative colitis and Crohn's disease) |
|
|
Nonmyeloid malignancies: |
Hodgkin disease, non-Hodgkin lymphomas, acute lymphoblastic leukemia, T cell lymphomas |
|
Nonhematological malignancies |
Solid tumors like carcinomas arising from lung, GIT, hepatobiliary system, thyroid, and genitourinary system[15] |
|
Other rare causes |
Graft versus host disease,[17] adrenal insufficiency,[18] atheroembolic disease,[19] Gleich syndrome (episodic angioedema with eosinophilia (EAE)[20] |

| Figure 2:The peripheral blood and bone marrow findings from a 56-year-old male patient who presented with paraparesis. Imaging showed a paraspinal mass; and subsequent fine-needle aspiration cytology confirmed a diagnosis of myeloid sarcoma. Hemogram showed leukocytosis (32 × 10^9/L) with eosinophilia (23%), mild anemia (hemoglobin 11 g/dL), and normal platelet counts (240 × 10^9/L). Peripheral blood film (A) showed eosinophils with bilobed and trilobed nuclei and cytoplasmic vacuoles (40x, May Grunwald Giemsa stain); Bone marrow aspirate (B) was hypercellular and showed excess of eosinophil precursors and eosinophils (40x, May Grunwald Giemsa stain); Bone marrow biopsy (C) showed interstitial excess of eosinophils (20x, hematoxylin and eosin stain) and reticulin stain (D) for mast cell tryptase highlighted the scattered as well as loose aggregates of mast cells (20x). Fluorescent in-situ hybridization (Vysis PDGFRA, CHIC2, FIP1L1 tricolor rearrangement probe, Abbott molecular, Illinois, United States) showed CHIC2 deletion (orange) (E) showed 3+ paratrabecular as well as interstitial fibrosis (20x). Immunohistochemistry (F) in 70%-cells consistent with FIP1L1::PDGFRA translocation. A diagnosis of myeloid neoplasm with eosinophilia and FIP1L1::PDGFRA translocation was made; and the patient responded to 100mg imatinib.
The diagnostic evaluation includes detailed medical history and physical examination, PBF examination, investigations to exclude common reactive/secondary causes of HE, and imaging to assess organ involvement. The importance of these is highlighted in [Table 2]. Because of the wide spectrum of infections associated with HE/HES and the nonavailability of complete infectious disease workup in most situations, it may not be possible to demonstrate the exact infectious agent, especially helminths. In such a situation, an empirical antihelminthic medication may be given; and further workup done in patients without a response.[7] [8] In view of the high incidence of parasitic infections, it is essential to understand that a positive serology may indicate exposure in the past and not necessarily the cause of HE.
|
Medical history |
To identify the cause and recognize any organ damage |
|---|---|
|
− Symptoms related to allergic disorders and family history of allergy |
HES secondary to allergy |
|
− Poor socioeconomic situation, poor nutrition and hygiene − Recent travel to tropical or endemic areas, exposure to pets, intake of undercooked meat or exotic dishes − Accidental ingestion/exposure to excreta of pets/insects − Drug intake (including indigenous drugs) − Recurrent infections with or without skin manifestations in children − Rheumatological/connective tissue disorders − Cough, breathlessness, pleuritic pain, abdominal pain, diarrhea, and neurological symptoms |
Increased risk of parasitic infections and infestations Increased risk of parasitic infections and infestations Increased risk of parasitic infections and infestations HES secondary to drugs including drug reaction with eosinophilia and systemic symptoms (DRESS) Possibility of primary immunodeficiency disorders with HE HE/HES secondary to rheumatological disorders May indicate organ damage due to HES |
|
Physical examination |
To identify the cause and recognize any organ damage |
|
− Pallor |
May indicate gastrointestinal helminthic infection |
|
− Moderate-to-massive splenomegaly |
May indicate clonal HE/myeloproliferative neoplasm with HE |
|
− Significant lymphadenopathy |
May indicate lymphoproliferative neoplasm or leukemia |
|
− Eczema |
Possibility of HE/HES secondary to allergy |
|
− Skin plaques/nodules |
Possibility of HE secondary to lymphoma |
|
− Pulmonary findings |
May indicate allergic bronchopulmonary aspergillosis or tropical pulmonary eosinophilia or HE-induced organ damage |
|
Investigations to exclude common reactive/secondary causes of HE |
|
|
− Fresh stool microscopy for ova, cysts, and parasites − Serological tests for parasites − Serological tests for viruses − ANA, C-ANCA, P-ANCA, anti-Rho/La, anti-smith, and other autoimmune serological markers − IgE levels − Aspergillus specific IgE − Serum LDH − Serum vitamin B12 − Day/night blood smears − Muscle biopsy − Microscopy of sputum or bronchoalveolar lavage fluid and fungal culture of respiratory samples |
Exclude parasitic infections Exclude microfilaria, trichinella, toxoplasma, Toxocara, cysticercus, hydatid, amoeba Exclude HIV, HBV, HCV Exclude autoimmune disorders and vasculitis High IgE in children may suggest reactive HE but may point towards hyper-IgE syndrome Exclude/diagnose aspergillosis High LDH may indicate lymphoproliferative disorder High vitamin B12 may indicate myeloproliferative disorder Exclude/diagnose filariasis Exclude/diagnosis trichinellosis Exclude/diagnosis fungal infections |
|
Other investigations − Chest X-ray − Computed tomography, − Electrocardiography (lungs and abdomen), − Pulmonary function test − Serum troponin T |
To assess organ involvement especially cardiac and lung Imaging gives us a clue about the cause of HE (e.g., lymphomas, solid malignancies, parasitic infections—cysticercosis, hydatid cyst) |
|
Abnormality |
Morphology |
|---|---|
|
RUNX1::RUNX1T1, CBFB: MYH11 |
AML |
|
BCR::ABL1 |
CML, AML, MPAL, B-ALL |
|
PDGFRA rearrangement (partners: FIP1L1, ETV6, BCR, FOXP1, STRN, TNKS2, KIF5B, SPECC1L, etc.) |
Isolated HE, MPN, MDS/MPN, blast phase of myeloid or lymphoid lineage (with eosinophilia ± neutrophilia, basophilia) |
|
PDGFRB rearrangement (partners: ETV6, PDE4DIP, WDR48, TPM3, SPTBN1, GOLGA4/B1, PRKG2, TNIP1, CEP85L, HIP1, BIN2, etc.) |
Isolated HE, MDS/MPN, MPN, blast phase of myeloid or lymphoid lineage (with eosinophilia) |
|
FGFR1 rearrangement (partners: ZMYM2, BCR, TPR, CEP43, CNTRL, CEP43G, etc.) |
B-ALL, T-ALL, MPAL, MPN, MPN/MDS (with eosinophilia ± neutrophilia, basophilia, monocytosis, erythrocytosis depending on the partner gene) |
|
JAK2 rearrangement (partners: PCM1, ETV6, BCR, etc.) |
MPN, MPN/MDS with eosinophilia and/or monocytosis |
|
FLT3 rearrangement (partners: ETV6, BCR, ZMYM2, TRIP11, SPTBN1, GOLGB1, CCDC88C, ZBTB44, and MYO18A) |
MDS, MPNs, MDS/MPNs, AML, B-ALL, T-ALL with eosinophilia. Myeloid sarcoma |
|
ETV6: ABL1 fusion |
CML like picture |
|
KIT p.D816V, JAK2 p.V617F, JAK2 exon 13 indels, STAT5B p.N642H,[43] DNMT3A, ASXL1, TET2, EZH2, SRSF2, SETBP1, CBL [a] |
CEL, SM associated with eosinophilia, PV with eosinophilia |
|
+8, -7, isochromosome 17, complex karyotype, 13q, 20q del, 1q abnormalities |
CEL |
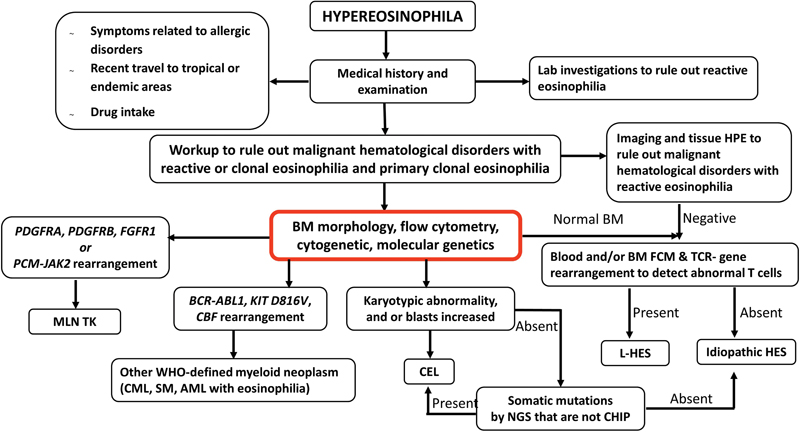
| Figure 3:Algorithmic approach in the diagnosis of hypereosinophilia/hypereosinophilic syndrome. AML, acute myeloid leukemia; BM, bone marrow; CBF, core binding factor; CEL, chronic eosinophilic leukemia; CHIP, clonal hematopoiesis of indeterminate potential; CML, chronic myeloid leukemia; FCM, flow cytometry; L-HES, lymphocytic variant of hypereosinophilia; MLN TK, myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions; NGS, next-generation sequencing; SM, systemic mastocytosis.
Conclusion
Considering the complex etiopathologies, the cost of testing, and the time involved, the workup needs to be tailored according to the urgency of the situation and the resources available. In urgent situations with organ damage, it is crucial to initiate appropriate management without waiting for the results of investigations. In contrast, in a resource-limited situation, it is acceptable to employ step-by-step rather than comprehensive testing to rule out the most common causes first.
Conflict of Interest
None declared.
Acknowledgement
The manuscript has been read and approved by all the authors, that the requirements for authorship have been met, and that each author believes that the manuscript represents honest work.
Authors' Contributions
D.S. wrote initial manuscript; S.S. revised and approved the final manuscript.
References
- 1 Valent P, Klion AD, Horny HP. et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130 (03) 607-612.e9
- 2 McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. J Exp Med 2002; 195 (11) F43-F47
- 3 Leru PM. Eosinophilic disorders: evaluation of current classification and diagnostic criteria, proposal of a practical diagnostic algorithm. Clin Transl Allergy 2019; 9: 36
- 4 Kato M, Kephart GM, Talley NJ. et al. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec 1998; 252 (03) 418-425
- 5 Simon HU, Rothenberg ME, Bochner BS. et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 2010; 126 (01) 45-49
- 6 Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol 2022; 97 (01) 129-148
- 7 Butt NM, Lambert J, Ali S. et al; British Committee for Standards in Haematology. Guideline for the investigation and management of eosinophilia. Br J Haematol 2017; 176 (04) 553-572
- 8 Sreedharanunni S, Varma N, Sachdeva MUS. et al. The spectrum of hypereosinophilia and associated clonal disorders - a real-world data based on combined retrospective and prospective analysis from a tropical setting. Mediterr J Hematol Infect Dis 2018; 10 (01) e2018052
- 9 Banday AZ, Bhattarai D, Bhagat N, Sreedharanunni S, Khurana S, Suri D. Pediatric hypereosinophilia and toxoplasma: peregrination beyond facileness. J Family Med Prim Care 2021; 10 (09) 3511-3514
- 10 Navabi B, Upton JEM. Primary immunodeficiencies associated with eosinophilia. Allergy Asthma Clin Immunol 2016; 12: 27
- 11 Long H, Zhang G, Wang L, Lu Q. Eosinophilic skin diseases: a comprehensive review. Clin Rev Allergy Immunol 2016; 50 (02) 189-213
- 12 Vignesh P, Kishore J, Kumar A. et al. A young child with eosinophilia, rash, and multisystem illness: drug rash, eosinophilia, and systemic symptoms syndrome after receipt of fluoxetine. Pediatr Dermatol 2017; 34 (03) e120-e125
- 13 Musette P, Janela B. New insights into drug reaction with eosinophilia and systemic symptoms pathophysiology. Front Med (Lausanne) 2017; 4: 179
- 14 Kardaun SH, Sekula P, Valeyrie-Allanore L. et al; RegiSCAR study group. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol 2013; 169 (05) 1071-1080
- 15 Zalewska E, Obołończyk Ł, Sworczak K. Hypereosinophilia in solid tumors-case report and clinical review. Front Oncol 2021; 11: 639395
- 16 Hu Z, Wang W, Thakral B. et al. Lymphocytic variant of hypereosinophilic syndrome: a report of seven cases from a single institution. Cytometry B Clin Cytom 2021; 100 (03) 352-360
- 17 Desmond A, Kilmer E, Cerny J, Nath R, Ramanathan M. Peripheral blood eosinophilia as a marker of chronic graft versus host disease (cGVHD). Biol Blood Marrow Transplant 2016; 22: S409
- 18 Takayasu S, Mizushiri S, Watanuki Y. et al. Eosinophil counts can be a predictive marker of immune checkpoint inhibitor-induced secondary adrenal insufficiency: a retrospective cohort study. Sci Rep 2022; 12 (01) 1294
- 19 Cecioni I, Fassio F, Gori S, Giudizi MG, Romagnani S, Almerigogna F. Eosinophilia in cholesterol atheroembolic disease. J Allergy Clin Immunol 2007; 120 (06) 1470-1471 , author reply 1471
- 20 Khoury P, Herold J, Alpaugh A. et al. Episodic angioedema with eosinophilia (Gleich syndrome) is a multilineage cell cycling disorder. Haematologica 2015; 100 (03) 300-307
- 21 Montgomery ND, Dunphy CH, Mooberry M. et al. Diagnostic complexities of eosinophilia. Arch Pathol Lab Med 2013; 137 (02) 259-269
- 22 Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev 2021; 49: 100809
- 23 Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007; 27 (03) 389-413
- 24 Cools J, DeAngelo DJ, Gotlib J. et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348 (13) 1201-1214
- 25 Roche-Lestienne C, Lepers S, Soenen-Cornu V. et al. Molecular characterization of the idiopathic hypereosinophilic syndrome (HES) in 35 French patients with normal conventional cytogenetics. Leukemia 2005; 19 (05) 792-798
- 26 Wang SA. The diagnostic work-up of hypereosinophilia. Pathobiology 2019; 86 (01) 39-52
- 27 Roufosse F, Weller PF. Practical approach to the patient with hypereosinophilia. J Allergy Clin Immunol 2010; 126 (01) 39-44
- 28 Goasguen JE, Bennett JM, Bain BJ. et al; International Working Group on Morphology of MDS. The role of eosinophil morphology in distinguishing between reactive eosinophilia and eosinophilia as a feature of a myeloid neoplasm. Br J Haematol 2020; 191 (03) 497-504
- 29 Gauchan D, Joshi N, Gill AS. et al. Does an elevated serum vitamin B(12) level mask actual vitamin B(12) deficiency in myeloproliferative disorders?. Clin Lymphoma Myeloma Leuk 2012; 12 (04) 269-273
- 30 Sahu KK, Malhotra P, Khadwal A. et al. Hypereosinophilia in Acute Lymphoblastic Leukemia: Two Cases with Review of Literature. Indian J Hematol Blood Transfus 2015; 31 (04) 460-465
- 31 Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022; 36: 1703-1719
- 32 Kapatia G, Remani ASN, Naseem S, Parihar M, Sreedharanunni S. Myeloid neoplasm with t(8;22)(p11;q11): a mimicker of chronic myeloid leukaemia in blast crisis. Indian J Hematol Blood Transfus 2021; 37 (02) 334-336
- 33 Fournier B, Balducci E, Duployez N. et al. B-ALL With t(5;14)(q31;q32); IGH-IL3 rearrangement and eosinophilia: a comprehensive analysis of a peculiar IGH-rearranged B-ALL. Front Oncol 2019; 9: 1374
- 34 Sreedharanunni S, Sachdeva MUS, Sharma S. et al. Paratrabecular myelofibrosis and occult mastocytosis are strong morphological clues to suspect FIP1L1-PDGFRA translocation in hypereosinophilia. Indian J Hematol Blood Transfus 2020; 36 (02) 384-389
- 35 Bacher U, Schanz J, Braulke F, Haase D. Rare cytogenetic abnormalities in myelodysplastic syndromes. Mediterr J Hematol Infect Dis 2015; 7 (01) e2015034
- 36 Batanian JR, Slovak ML, Mohamed A, Dobin S, Luthardt FW, Keitges EA. Trisomy 15 is frequently observed as a minor clone in patients with Anemia/MDS/NHL and as a major clone in patients with AML. Cancer Genet Cytogenet 2000; 121 (02) 186-189
- 37 Sharma P, Rana S, Sreedharanunni S. et al. An evaluation of a fluorescence in situ hybridization strategy using air-dried blood and bone-marrow smears in the risk stratification of pediatric b-lineage acute lymphoblastic leukemia in resource-limited settings. J Pediatr Hematol Oncol 2021; 43 (04) e481-e485
- 38 Virk H, Rathore S, Anshu A. et al. Imatinib responsive erythrocytosis in a patient with FIP1L1:PDGFRA rearranged myeloid neoplasm with hypereosinophilia - Another manifestation of a stem cell neoplasm. Leuk Res 2022; 121: 106922
- 39 Olsson-Arvidsson L, Norberg A, Sjögren H, Johansson B. Frequent false-negative FIP1L1-PDGFRA FISH analyses of bone marrow samples from clonal eosinophilia at diagnosis. Br J Haematol 2020; 188 (05) e76-e79
- 40 Wang SA, Tam W, Tsai AG. et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol 2016; 29 (08) 854-864
- 41 Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica 2009; 94 (09) 1236-1241
- 42 Morsia E, Reichard K, Pardanani A, Tefferi A, Gangat N. WHO defined chronic eosinophilic leukemia, not otherwise specified (CEL, NOS): a contemporary series from the Mayo Clinic. Am J Hematol 2020; 95 (07) E172-E174
- 43 Sreedharanunni S, Jamwal M, Balakrishnan A. et al. Chronic eosinophilic leukemia with recurrent STAT5B N642H mutation-an entity with features of myelodysplastic syndrome/ myeloproliferative neoplasm overlap. Leuk Res 2022; 112: 106753
- 44 Klion AD. How I treat hypereosinophilic syndromes. Blood 2015; 126 (09) 1069-1077
Address for correspondence
Publication History
Article published online:
17 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig 1 : The process of eosinophilopoiesis.
| Figure 2:The peripheral blood and bone marrow findings from a 56-year-old male patient who presented with paraparesis. Imaging showed a paraspinal mass; and subsequent fine-needle aspiration cytology confirmed a diagnosis of myeloid sarcoma. Hemogram showed leukocytosis (32 × 10^9/L) with eosinophilia (23%), mild anemia (hemoglobin 11 g/dL), and normal platelet counts (240 × 10^9/L). Peripheral blood film (A) showed eosinophils with bilobed and trilobed nuclei and cytoplasmic vacuoles (40x, May Grunwald Giemsa stain); Bone marrow aspirate (B) was hypercellular and showed excess of eosinophil precursors and eosinophils (40x, May Grunwald Giemsa stain); Bone marrow biopsy (C) showed interstitial excess of eosinophils (20x, hematoxylin and eosin stain) and reticulin stain (D) for mast cell tryptase highlighted the scattered as well as loose aggregates of mast cells (20x). Fluorescent in-situ hybridization (Vysis PDGFRA, CHIC2, FIP1L1 tricolor rearrangement probe, Abbott molecular, Illinois, United States) showed CHIC2 deletion (orange) (E) showed 3+ paratrabecular as well as interstitial fibrosis (20x). Immunohistochemistry (F) in 70
| Figure 3:Algorithmic approach in the diagnosis of hypereosinophilia/hypereosinophilic syndrome. AML, acute myeloid leukemia; BM, bone marrow; CBF, core binding factor; CEL, chronic eosinophilic leukemia; CHIP, clonal hematopoiesis of indeterminate potential; CML, chronic myeloid leukemia; FCM, flow cytometry; L-HES, lymphocytic variant of hypereosinophilia; MLN TK, myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions; NGS, next-generation sequencing; SM, systemic mastocytosis.
References
- 1 Valent P, Klion AD, Horny HP. et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130 (03) 607-612.e9
- 2 McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. J Exp Med 2002; 195 (11) F43-F47
- 3 Leru PM. Eosinophilic disorders: evaluation of current classification and diagnostic criteria, proposal of a practical diagnostic algorithm. Clin Transl Allergy 2019; 9: 36
- 4 Kato M, Kephart GM, Talley NJ. et al. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec 1998; 252 (03) 418-425
- 5 Simon HU, Rothenberg ME, Bochner BS. et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 2010; 126 (01) 45-49
- 6 Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol 2022; 97 (01) 129-148
- 7 Butt NM, Lambert J, Ali S. et al; British Committee for Standards in Haematology. Guideline for the investigation and management of eosinophilia. Br J Haematol 2017; 176 (04) 553-572
- 8 Sreedharanunni S, Varma N, Sachdeva MUS. et al. The spectrum of hypereosinophilia and associated clonal disorders - a real-world data based on combined retrospective and prospective analysis from a tropical setting. Mediterr J Hematol Infect Dis 2018; 10 (01) e2018052
- 9 Banday AZ, Bhattarai D, Bhagat N, Sreedharanunni S, Khurana S, Suri D. Pediatric hypereosinophilia and toxoplasma: peregrination beyond facileness. J Family Med Prim Care 2021; 10 (09) 3511-3514
- 10 Navabi B, Upton JEM. Primary immunodeficiencies associated with eosinophilia. Allergy Asthma Clin Immunol 2016; 12: 27
- 11 Long H, Zhang G, Wang L, Lu Q. Eosinophilic skin diseases: a comprehensive review. Clin Rev Allergy Immunol 2016; 50 (02) 189-213
- 12 Vignesh P, Kishore J, Kumar A. et al. A young child with eosinophilia, rash, and multisystem illness: drug rash, eosinophilia, and systemic symptoms syndrome after receipt of fluoxetine. Pediatr Dermatol 2017; 34 (03) e120-e125
- 13 Musette P, Janela B. New insights into drug reaction with eosinophilia and systemic symptoms pathophysiology. Front Med (Lausanne) 2017; 4: 179
- 14 Kardaun SH, Sekula P, Valeyrie-Allanore L. et al; RegiSCAR study group. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol 2013; 169 (05) 1071-1080
- 15 Zalewska E, Obołończyk Ł, Sworczak K. Hypereosinophilia in solid tumors-case report and clinical review. Front Oncol 2021; 11: 639395
- 16 Hu Z, Wang W, Thakral B. et al. Lymphocytic variant of hypereosinophilic syndrome: a report of seven cases from a single institution. Cytometry B Clin Cytom 2021; 100 (03) 352-360
- 17 Desmond A, Kilmer E, Cerny J, Nath R, Ramanathan M. Peripheral blood eosinophilia as a marker of chronic graft versus host disease (cGVHD). Biol Blood Marrow Transplant 2016; 22: S409
- 18 Takayasu S, Mizushiri S, Watanuki Y. et al. Eosinophil counts can be a predictive marker of immune checkpoint inhibitor-induced secondary adrenal insufficiency: a retrospective cohort study. Sci Rep 2022; 12 (01) 1294
- 19 Cecioni I, Fassio F, Gori S, Giudizi MG, Romagnani S, Almerigogna F. Eosinophilia in cholesterol atheroembolic disease. J Allergy Clin Immunol 2007; 120 (06) 1470-1471 , author reply 1471
- 20 Khoury P, Herold J, Alpaugh A. et al. Episodic angioedema with eosinophilia (Gleich syndrome) is a multilineage cell cycling disorder. Haematologica 2015; 100 (03) 300-307
- 21 Montgomery ND, Dunphy CH, Mooberry M. et al. Diagnostic complexities of eosinophilia. Arch Pathol Lab Med 2013; 137 (02) 259-269
- 22 Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev 2021; 49: 100809
- 23 Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007; 27 (03) 389-413
- 24 Cools J, DeAngelo DJ, Gotlib J. et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348 (13) 1201-1214
- 25 Roche-Lestienne C, Lepers S, Soenen-Cornu V. et al. Molecular characterization of the idiopathic hypereosinophilic syndrome (HES) in 35 French patients with normal conventional cytogenetics. Leukemia 2005; 19 (05) 792-798
- 26 Wang SA. The diagnostic work-up of hypereosinophilia. Pathobiology 2019; 86 (01) 39-52
- 27 Roufosse F, Weller PF. Practical approach to the patient with hypereosinophilia. J Allergy Clin Immunol 2010; 126 (01) 39-44
- 28 Goasguen JE, Bennett JM, Bain BJ. et al; International Working Group on Morphology of MDS. The role of eosinophil morphology in distinguishing between reactive eosinophilia and eosinophilia as a feature of a myeloid neoplasm. Br J Haematol 2020; 191 (03) 497-504
- 29 Gauchan D, Joshi N, Gill AS. et al. Does an elevated serum vitamin B(12) level mask actual vitamin B(12) deficiency in myeloproliferative disorders?. Clin Lymphoma Myeloma Leuk 2012; 12 (04) 269-273
- 30 Sahu KK, Malhotra P, Khadwal A. et al. Hypereosinophilia in Acute Lymphoblastic Leukemia: Two Cases with Review of Literature. Indian J Hematol Blood Transfus 2015; 31 (04) 460-465
- 31 Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022; 36: 1703-1719
- 32 Kapatia G, Remani ASN, Naseem S, Parihar M, Sreedharanunni S. Myeloid neoplasm with t(8;22)(p11;q11): a mimicker of chronic myeloid leukaemia in blast crisis. Indian J Hematol Blood Transfus 2021; 37 (02) 334-336
- 33 Fournier B, Balducci E, Duployez N. et al. B-ALL With t(5;14)(q31;q32); IGH-IL3 rearrangement and eosinophilia: a comprehensive analysis of a peculiar IGH-rearranged B-ALL. Front Oncol 2019; 9: 1374
- 34 Sreedharanunni S, Sachdeva MUS, Sharma S. et al. Paratrabecular myelofibrosis and occult mastocytosis are strong morphological clues to suspect FIP1L1-PDGFRA translocation in hypereosinophilia. Indian J Hematol Blood Transfus 2020; 36 (02) 384-389
- 35 Bacher U, Schanz J, Braulke F, Haase D. Rare cytogenetic abnormalities in myelodysplastic syndromes. Mediterr J Hematol Infect Dis 2015; 7 (01) e2015034
- 36 Batanian JR, Slovak ML, Mohamed A, Dobin S, Luthardt FW, Keitges EA. Trisomy 15 is frequently observed as a minor clone in patients with Anemia/MDS/NHL and as a major clone in patients with AML. Cancer Genet Cytogenet 2000; 121 (02) 186-189
- 37 Sharma P, Rana S, Sreedharanunni S. et al. An evaluation of a fluorescence in situ hybridization strategy using air-dried blood and bone-marrow smears in the risk stratification of pediatric b-lineage acute lymphoblastic leukemia in resource-limited settings. J Pediatr Hematol Oncol 2021; 43 (04) e481-e485
- 38 Virk H, Rathore S, Anshu A. et al. Imatinib responsive erythrocytosis in a patient with FIP1L1:PDGFRA rearranged myeloid neoplasm with hypereosinophilia - Another manifestation of a stem cell neoplasm. Leuk Res 2022; 121: 106922
- 39 Olsson-Arvidsson L, Norberg A, Sjögren H, Johansson B. Frequent false-negative FIP1L1-PDGFRA FISH analyses of bone marrow samples from clonal eosinophilia at diagnosis. Br J Haematol 2020; 188 (05) e76-e79
- 40 Wang SA, Tam W, Tsai AG. et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol 2016; 29 (08) 854-864
- 41 Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica 2009; 94 (09) 1236-1241
- 42 Morsia E, Reichard K, Pardanani A, Tefferi A, Gangat N. WHO defined chronic eosinophilic leukemia, not otherwise specified (CEL, NOS): a contemporary series from the Mayo Clinic. Am J Hematol 2020; 95 (07) E172-E174
- 43 Sreedharanunni S, Jamwal M, Balakrishnan A. et al. Chronic eosinophilic leukemia with recurrent STAT5B N642H mutation-an entity with features of myelodysplastic syndrome/ myeloproliferative neoplasm overlap. Leuk Res 2022; 112: 106753
- 44 Klion AD. How I treat hypereosinophilic syndromes. Blood 2015; 126 (09) 1069-1077