 PDF
PDF  Views
Views  Share
Share


Inflammatory myofibroblastic tumor of kidney with heterotopic bone formation: An unusual case mimicking a renal malignancy
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2013; 34(04): 320-322
DOI: DOI: 10.4103/0971-5851.125257
Abstract
Inflammatory myofibroblastic tumor (IMFT) or pseudotumor is a rare tumor that can be seen in various organs. It is an enigmatic tumor of unknown etiology and difficult to differentiate from malignancy, clinically as well as radiologically. Histologically, it is composed of variable proportions of proliferating spindle myofibroblasts, fibroblasts, extracellular collagen, lymphocytes and plasma cells. This mainly affects the urinary bladder or prostate. Renal involvement is extremely rare and may pose diagnostic difficulty to the clinician. We describe a renal IMFT, mimicking renal malignancy, having unusual histological features.
Publication History
Article published online:
19 July 2021
© 2013. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Inflammatory myofibroblastic tumor (IMFT) or pseudotumor is a rare tumor that can be seen in various organs. It is an enigmatic tumor of unknown etiology and difficult to differentiate from malignancy, clinically as well as radiologically. Histologically, it is composed of variable proportions of proliferating spindle myofibroblasts, fibroblasts, extracellular collagen, lymphocytes and plasma cells. This mainly affects the urinary bladder or prostate. Renal involvement is extremely rare and may pose diagnostic difficulty to the clinician. We describe a renal IMFT, mimicking renal malignancy, having unusual histological features.
INTRODUCTION
Inflammatory myofibroblastic tumor (IMFT), also called as inflammatory pseudotumor was an entity originally described in lung. It has now been identified at multiple extrapulmonary anatomical sites viz. gastrointestinal tract, soft-tissue, orbit, spleen and lymph nodes.[1] In the urinary tract, it most commonly occurs in the urinary bladder.[2] Renal inflammatory pseudotumor is extremely rare with less than 25 cases reported in English literature.[3,4] Histologically, it is a tumor composed of differentiated myofibroblastic spindle cells and usually numerous plasma cells and/or lymphocytes. The literature until recently was mired in controversy regarding the nature of these myofibroblastic cells. However, with the advent of molecular techniques it has been proved that a subset of these tumors is neoplastic in nature, harboring translocations of activin receptor-like kinase-1 (ALK-1) gene.[5] We report a rare case of renal IMFT that radiologically mimicked a malignancy and had unusual histological features.
CASE REPORT
A 32-year-old male patient came with complaints of pain in the lower abdomen and urinary tract symptoms since 1 year. The patient had a history of renal lithiasis 8 years back. Laboratory investigations including complete blood count, serum biochemistry and urine analysis were within the normal limits. Computed tomography (CT) scan revealed a heterogeneously enhancing mass lesion measuring 4.5 cm × 4.4 cm arising from the lower pole of the right kidney with an exophytic component. Calcific areas were also noted in the mass. It was confined to the cortex with no evidence of extension into renal calyces. Right renal vein was patent and there was no evidence of renal vein thrombosis. The contralateral kidney was normal. Radiological and clinical features strongly suggested a renal cell carcinoma (RCC) for which the patient underwent a right radical nephrectomy. Macroscopically, a well-circumscribed hard grey white tumor was noted in the lower pole of nephrectomy specimen with exophytic component also. The tumor was hard and had a gritty sensation on cutting through it. Histological examination revealed a tumor composed of cellular and hypocellular hyalinized areas. The cellular foci were composed of spindle cells in short fascicles and prominent storiform pattern. Spindle cells showed mild atypia and mitotic activity ranged from 4 to 7/10 high power field (HPF). There were lymphoid aggregates with germinal center formation and plasma cells interspersed amongst these spindle cells. Extravasated red blood cells were also seen prominently at many foci. Within the hypocellular hyalinized zones, the spindle cells were arranged in an unusual perivascular whorled pattern resembling meningeal whorls. In addition, heterotopic bone formation was seen bordered by osteoblastic cells [Figure 1]. Mitotic rate up to 3-6/10 HPF was appreciated focally in the cellular areas. However, no atypical mitosis was appreciated. No areas of conventional RCC were demonstrable even after studying additional sections from the tumor. On immunohistochemistry, the spindle cells were strongly positive for smooth muscle antibody [Figure 2] and were negative for CD 34, ALK-1, CK7, CD 23, CD21 and h-caldesmon. p53 was positive in few of the spindle cells. The remaining renal parenchyma was unremakable. Surgical margins of excision were clear with no evidence of extension into the renal vein or renal pelvis. The patient is asymptomatic 18 months after his surgery.
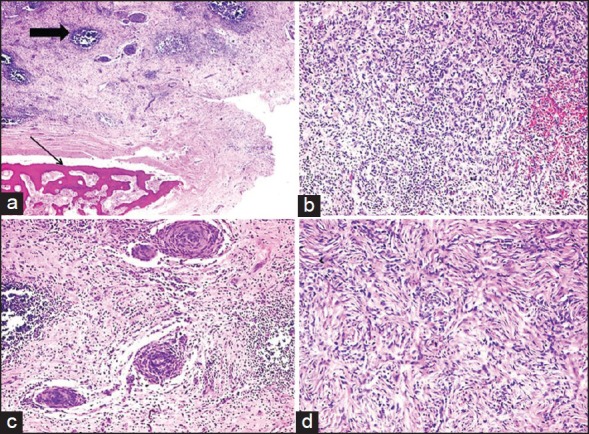
| Fig. 1 Low power view of inflammatory myofibroblastic tumor (IMFT) of kidney with heterotopic bone formation (thin arrow). Even at this power the lymphoid aggregates are prominent (thick arrow), (a) (H and E, ×40). The presence of spindle cells, inflammatory cells with extravasated red blood cells, (b) (H and E, ×100), perivascular whorls of spindle cells, (c) (H and E, ×100) and focal storiform pattern, (d) (H and E, ×200) was demonstrable in various areas of IMFT
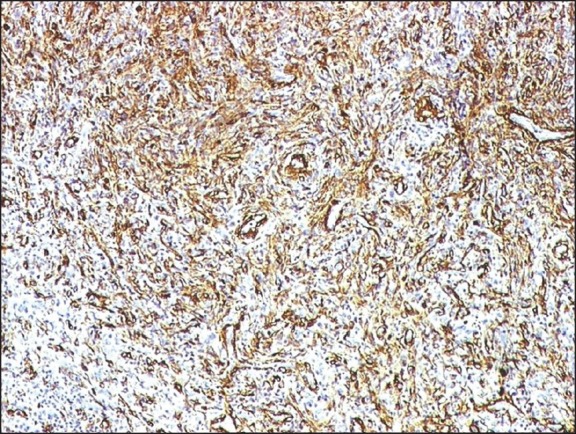
| Fig. 2 The spindle cells of inflammatory myofibroblastic tumor kidney strongly expressed smooth muscle actin (smooth muscle antibody, indirect immunoperoxidase, ×100)
DISCUSSION
IMFT is a rare entity in the genitourinary tract with most reported cases occurring in the urinary bladder. The clinical presentation varies depending upon the anatomical sites. Radiologically, the findings are non-specific and only reveal a solid well-defined mass. IMFT has been previously known under various synonyms such as inflammatory pseudotumor, plasma cell granuloma, inflammatory myofibrohistiocytic proliferation, pseudosarcomatous myofibroblastic proliferation and xanthomatous pseudotumor. This confusion in nomenclature arose due to ambiguity regarding its origin, as to whether it is a reactive process or a true neoplasm. It was proposed that the inflammatory reaction may be secondary to surgery, trauma or infections implicating epstein-barr virus,[7] human herpes virus 8.[8] There are studies identifying clonal abnormalities of the short arm of chromosome 2 in the region of p21-23 in myofibroblastic tumors, the site of ALK gene that encodes for a tyrosine kinase receptor. ALK abnormalities have been reported in 37-60% of the cases studied, by immunohistochemistry and fluorescence in-situ hybridization.[5] ALK negative cases tend to occur in older patients and ALK positive cases tend to occur in younger patients (children and younger adults).[3] This is significant owing to the fact that ALK was negative in our adult patient.
Histologically, three patterns have been elucidated in these tumors, including a myxoid vascular pattern, a compact spindle cell pattern, and a hypocellular fibrous pattern (nodular fasciitis like, fibrous histiocytoma like and a desmoid or scar tissue.[5,6] In a given case, there may be admixture or predominance of any of these patterns. Our case had a predominance of fibrous histiocytoma-like as well as desmoid-like hypocellular areas. The presence of perivascular whorls of spindle cells was striking in our case. Heterotopic bone formation rarely been seen in IMFT.[5,6]
The differential diagnoses of this entity are sarcomatous change in a RCC, inflammatory fibrosarcomas, low grade neurogenic tumor, dendritic cell tumors and benign tumors like angiomyolipoma and xanthogranulomatous pyelonephritis.[9] Most important from a clinical perspective is to differentiate IMFT from a sarcomatoid RCC. The absence of significant nuclear pleomorphism in the spindle cell component, absence of a biphasic appearance with another typical pattern of renal-cell carcinoma, and lack of evidence for epithelial differentiation by immunohistochemistry were against an interpretation of sarcomatoid RCC in our case.[10,11]
Biological behavior of IMFT is difficult to predict. Local recurrences and malignant transformation have been reported in a small subset of patients, especially in those in whom complete surgical excision has not been possible.[5] The factors associated with aggressive behavior with recurrences or malignant transformations are cellular atypia, ganglion like cells, p53 expression, and deoxyribonucleic acid aneuploidy.[3] In view of the focally increased mitosis and p53 positivity the biological behavior regarding recurrence is difficult to ascertain in our case. However, evidence points that IMFT of kidney has no potential for recurrence or metastases.[3] Nevertheless, such cases have to be closely followed-up since there is evidence in the literature demonstrating association of RCC arising in a background of inflammatory pseudotumor of kidney.[11]
In conclusion, IMFT of renal parenchyma is a proliferative lesion of myofibroblasts of uncertain pathogenesis. In our case, since patient had a long history of urinary tract symptoms, we surmise chronic inflammation as a pathogenetic event. Complete surgical excision is the mainstay of treatment.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
| Fig. 1 Low power view of inflammatory myofibroblastic tumor (IMFT) of kidney with heterotopic bone formation (thin arrow). Even at this power the lymphoid aggregates are prominent (thick arrow), (a) (H and E, ×40). The presence of spindle cells, inflammatory cells with extravasated red blood cells, (b) (H and E, ×100), perivascular whorls of spindle cells, (c) (H and E, ×100) and focal storiform pattern, (d) (H and E, ×200) was demonstrable in various areas of IMFT
| Fig. 2 The spindle cells of inflammatory myofibroblastic tumor kidney strongly expressed smooth muscle actin (smooth muscle antibody, indirect immunoperoxidase, ×100)