 PDF
PDF  Views
Views  Share
Share


Imatinib mesylate resistance and mutations: An Indian experience
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2013; 34(03): 213-220
DOI: DOI: 10.4103/0971-5851.123748
Abstract
The treatment of chronic myeloid leukemia (CML) has been revolutionized by the small molecule selective kinase inhibitor imatinib mesylate. Imanitib was the first BCR-ABL targeted agent approved for the treatment of CML patients and confers significant response in most patients; however, a substantial number of patients are initially refractory to the drug or may develop resistance during the course of treatment. Point mutations in the kinase domain (KD) of BCR-ABL that impact drug binding have been identified as one of the major mechanisms of resistance. We present here an overview of the current practice in monitoring for such mutations, including the methods used, criteria for investigating and guidelines for reporting the mutations. We further present and discuss the experience of our own laboratory in studying the KD mutations in Indian CML patients on imatinib treatment.
Publication History
Article published online:
19 July 2021
© 2013. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
The treatment of chronic myeloid leukemia (CML) has been revolutionized by the small molecule selective kinase inhibitor imatinib mesylate. Imanitib was the first BCR-ABL targeted agent approved for the treatment of CML patients and confers significant response in most patients; however, a substantial number of patients are initially refractory to the drug or may develop resistance during the course of treatment. Point mutations in the kinase domain (KD) of BCR-ABL that impact drug binding have been identified as one of the major mechanisms of resistance. We present here an overview of the current practice in monitoring for such mutations, including the methods used, criteria for investigating and guidelines for reporting the mutations. We further present and discuss the experience of our own laboratory in studying the KD mutations in Indian CML patients on imatinib treatment.
INTRODUCTION
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia chromosome, which results from a balanced genetic translocation between BCR gene on chromosome 22 and ABL gene on chromosome 9.[1] This translocation results in constitutive activation of the hybrid BCR-ABL gene that confers an anti-apoptotic advantage to the mutated cell and forms the pathophysiologic basis of CML.[2]
As per current practices, treatment of CML relies upon tyrosine kinase inhibitors (TKIs) directed against BCR-ABL. Imatinib mesylate (Gleevec®, Novartis Pharmaceuticals) was the first TKI approved for the treatment of CML and is the current first-line treatment. Approval of this drug was based on data from the International Randomized Study of Interferon (IFN) and STI571.[3] While most patients in this trial benefited from imatinib treatment, a significant number were found to be either initially refractory (primary resistance) or developed resistance during the course of treatment (secondary/acquired resistance). Some of these patients progressed to accelerated phase (AP) or blast phase CML, while others suffered a loss of previously established hematologic or major cytogenetic response (MCyR). It has been shown that secondary resistance may result in progression to advanced phases in 7% of patients and as relapsed disease in approximately 17% of patients on imatinib.[4] Importantly, the incidence of resistance to imatinib is higher in patients with more advanced stages of CML, with relapse occurring in most patients who initially respond to treatment.
MECHANISMS OF IMATINIB RESISTANCE DEVELOPMENT
BCR-ABL dependent resistance
Resistance is often categorized as BCR-ABL dependent or BCR-ABL independent. BCR-ABL dependent mechanisms are believed to be the most common reason for the development of resistance. Kinase domain (KD) mutations represent the most common mechanism of acquired resistance to imatinib, occurring in 30-50% of cases. The ABL tyrosine kinase moves between a catalytically active open conformation and an inactive closed one. Imatinib has been shown to bind to the ABL KD in the inactive, or closed, conformation and to induce a variety of conformational changes to the protein upon binding.[5] While some resistance-associated mutations occur at amino acid positions implicated in directly binding the drug, the majority are believed to prevent the KD from adopting the specific conformation to which imatinib binds.[6] The A-loop is a major regulator of BCR-ABL kinase activity by adopting either a closed (inactive) or open (active) conformation and A-loop mutations often destabilize the inactive conformation that is required for imatinib binding.
Till-date, more than 90 discrete resistance conferring point mutations at 57 residues in the ABL kinase have been documented.[7] These mutations generally fall within four regions of the KD: The adhenosine triphosphate (ATP)-binding loop (P-loop), the drug binding site, the catalytic site and the activation loop (A-loop). The most frequently occurring mutations (30-40%) associated with BCR-ABL are those of the P-loop.[8] P-loop mutations are more frequent in advanced chronic phase (CP) and AP or blast crisis (BC) phase CML and there is data to show that patients who experience P-loop mutations have a poor prognosis for response and survival.[9] Kinase assays have shown that P-loop mutations are 70-100 folds less sensitive to imatinib and have higher transforming activity than wild-type BCR-ABL.[10] Another frequently occurring mutation, T315I, also known as the “gatekeeper mutation” represents a key challenge in the treatment of CML as neither imatinib nor the two second-generation TKIs are active against it.[11] New treatment strategies are currently in development to address this problem. Mutations associated with secondary imatinib resistance occur more frequently in later stages of the disease and are associated with older age, prior IFN therapy, initiation of imatinib therapy in AP or BC, development of clonal evolution, high-risk Sokal score at diagnosis and failure to achieve complete cytogenetic response (CCyR) by 12 months.[12] Additional mechanisms of BCR-ABL dependent mechanisms of imatinib resistance include genomic amplification of BCR-ABL[13] and overexpression of BCR-ABL messenger ribonucleic acid (RNA).[14] Other mediators of imatinib resistance are overexpression of the plasma-protein alpha-1 acid glycoprotein and the cellular efflux protein P-glycoprotein.[15,16]
BCR-ABL independent resistance
Although most cases of acquired imatinib resistance are associated with reactivation of BCR-ABL activity through the mechanisms described above, there are cases of resistance that appear to occur through mechanisms independent of BCR-ABL. One such mechanism is the constitutive activation of downstream signaling molecules, which could result in the activation of the pathway regardless of BCR-ABL inhibition, thereby resulting in what would appear to be imatinib resistance. The Src family kinases (SFKs) are one such example of downstream signaling molecules. SFKs regulate cell proliferation and survival; have been implicated in the development of late-stage CML, as well as BCR-ABL-independent imatinib resistance.[17] In vitro studies have shown that CML cell lines exhibiting imatinib resistance unrelated to BCR-ABL overexpress LYN and HCK.[18]
DETECTION OF ABL KD MUTATIONS
It is now accepted that the expansion of a Ph-positive clone carrying an ABL KD mutation is associated with resistance to imatinib and in some cases precedes or accompanies progression to advanced-phase disease.[9] By corollary, this would mean that the KD mutations above a certain level should be identified as early as possible because they may indicate the need to reconsider the therapeutic strategy. Furthermore, the probability of finding a mutant clone is very low in a patient who has a stable or declining level of BCR-ABL transcripts. The incidence of mutations in imatinib-naive CP patients and patients in CCyR is usually also low.[19] Moreover, mutant clones at a low level may not necessarily have the same clinical significance as clones that are detected in the context of a rising disease burden and this has been shown even in case of the completely resistant T315I mutant.[20] A possible explanation is that only KD mutant clones originating in a leukemic stem cell are able to sustain malignant hematopoiesis, whereas mutants generated in more differentiated progenitor cells are passively phased-out over time. What this indicates in practical terms is that use of a highly sensitive technique for mutation detection before therapy might sometimes produce misleading results. However, the situation might be different if the mutant clone is dominant before imatinib treatment and is seen in patients with high risk CML.
Techniques used for KD mutation analysis
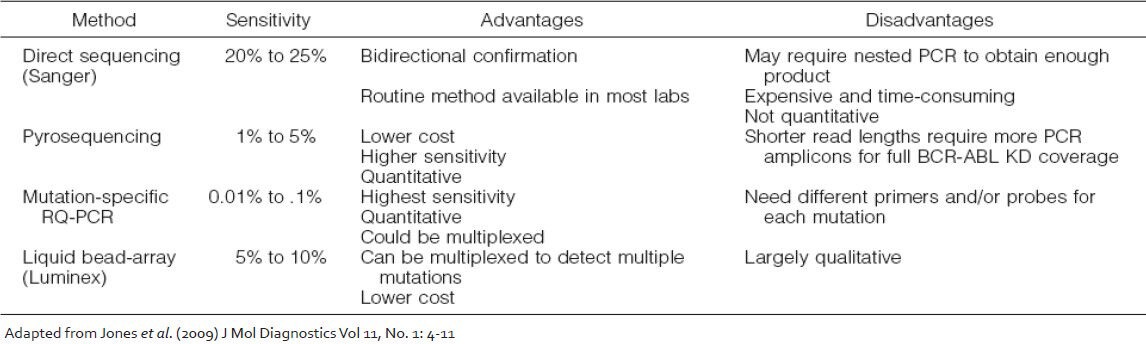
A number of technologies are currently available for detection and study of KD mutations which vary widely in their sensitivity, specificity and bias and the final choice of which method should be used is largely dependent on the clinical utility of the information they provide [Table 1]. The direct Sanger sequencing method has a sensitivity of 15-20%, whereas the highly sensitive mutation-specific quantitative polymerase chain reaction (PCR) methods can reliably detect a mutant transcript down to 1 in 10,000 BCR-ABL transcripts. Other screening methods for BCR-ABL KD mutations that have been reported include denaturing high performance liquid chromatography, targeted microarrays and liquid bead arrays. In all of these methods a final confirmation with direct sequencing is warranted for the abnormal cases. Several quantitative mutation detection methods that have been developed to track the proportion of a mutated clone after therapy switch[21] including PCR-based pyrosequencing and mutation-specific quantitative PCR are also becoming popular. However, since the detection of low levels of mutant clones may not be clinically significant, direct sequencing of the BCR-ABL transcript by the Sanger method is currently the most appropriate screening test and was recommended by an international consensus panel.[22]
Table 1
Techniques available for BCR-ABL kinase domain mutations analysis
When to perform KD mutation analysis: Correlation with response criteria on international scale (IS)
In a study carried out by Chu et al.[23] BCR-ABL KD mutations were identified in stem cells from patients of CML without clinical evidence of resistant disease. It therefore becomes important to address the question as to when should one perform KD mutation screening. An international consensus group was convened to develop guidelines for use of BCR-ABL transcript monitoring and mutation testing in CML. Their recommendations are published and are available as guidelines.[24] Updates to the same with incorporation of newer response criteria are now available.[25,26,27] Following these recommendations, KD mutation screening is recommended in cases of imatinib failure, suboptimal response or increasing BCR-ABL transcript level [Table 2]. Mutation screening is also recommended at the time of progression to AP or blast phase CML and when a switch to 2nd generation TKI is being considered. On the other hand, there have been suggestions that regular mutation screening on all patients should bed one because even some patients in stable CCyR carry mutations, the presence of which may precede a rise in BCR-ABL transcript value.[28] The study authors concluded that the systematic screening of all patients in CCyR might not be cost-effective and suggested that a reasonable compromise would be twice yearly mutation screen for all patients who still have BCR-ABL values >0.3% IS.
Table 2
Recommended frequencies of response assessment in chronic myeloid leukemia patients on imatinib
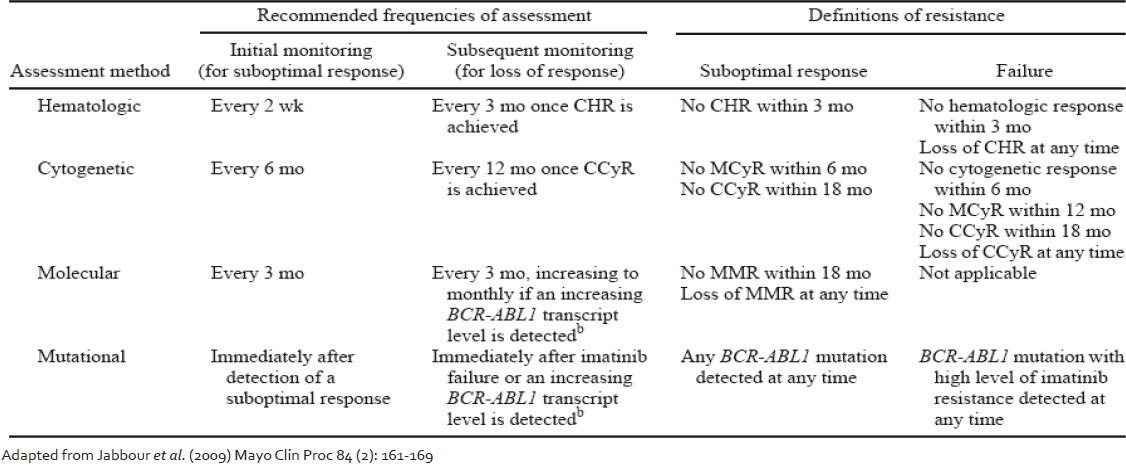
The present ELN guidelines[25] may also be interpreted in light of the BCR-ABL transcript values. The highest incidence of mutations has been reported in patients failing to achieve a MCyR by 6 months.[8] BCR-ABL values lower than 10% IS are approximately equivalent to the achievement of a MCyR.[29] Hughes and Branford[26] have recommended performing mutation analysis for all patients treated with imatinib who have a BCR-ABL value of higher than 10% IS at 6 months, followed by mutation screening every 3 months until the BCR-ABL value falls below 1% IS. In our own study (data not shown) we have found the non-achievement of 10% IS BCR-ABL transcript value to be an important parameter highly associated with the presence of a mutation. Mutation analysis is also indicated for patients who have a significant rise in BCR-ABL (2-5 folds) and have a confirmed loss of a major molecular response unless there is a clear association between the rising BCRABL level and a dose reduction or interruption.
Reporting and interpretation of KD mutations
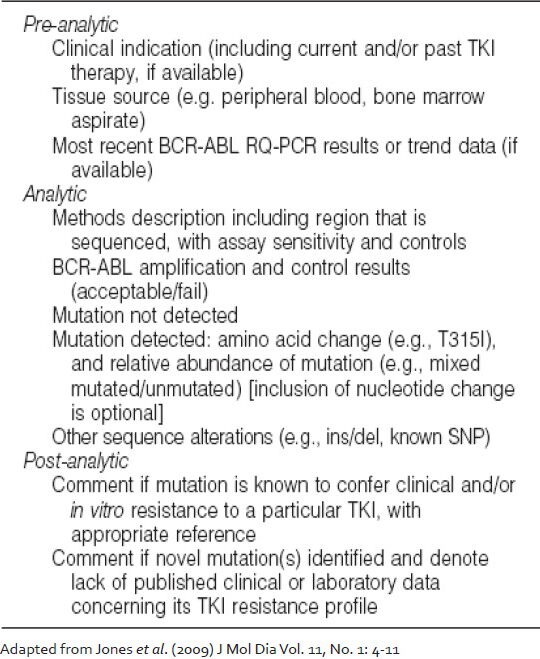
The reporting recommendations for BCR-ABL KD mutation analysis released by College of American Pathologists are summarized in Table 3. Briefly, the report should encompass elements from the pre-analytical, analytical and post-analytical processes. If a mutation is detected, it should be indicated according to standard amino acid substitution nomenclature with the nucleotide change clearly marked.[30] In case a non-quantitative mutation detection method such as Sanger sequencing is used, an estimate of the relative quantity of the mutation should ideally be provided, which would serve as a baseline value for subsequent monitoring. More than one mutation should be similarly reported. It is recommended that the clinical correlation of the finding should be clearly indicated; if novel mutations or other genetic findings (e.g., insertion/deletion events, known or previously unreported single nucleotide polymorphisms) are identified, a statement should be added indicating that the impact of the alteration on TKI resistance is not currently known. Other information that could be included in a report concern the clinical indications for testing (e.g., loss of BCR-ABL molecular response, cytogenetic relapse, etc.), the current TKI therapy and the most recent BCR-ABL transcript level. Although the ultimate goal of mutation testing is to guide therapeutic decisions, the report should not contain specific recommendations concerning, which therapies are optimal for any given patient. One possible pitfall of mutation screening is the detection of ABL polymorphisms that may be mistaken for mutations.[31,32] There are at least 10 known polymorphisms in the ABL KD, which are L140P (419 T > C), G159S (475 G > A), T240T (720 G > A), K247R (740 A > G), L298L (894 G > A), L354L (1062 G > A), T315T (945 T > G), E459K (1375 G > A), E499E (1497 A > G) and S520T (1559 G > C). None of these have a known effect on TKI binding. Hence, it is important to exclude the occurrence of polymorphism before reporting any novel mutations.
Table 3
Reporting recommendations for BCR-ABL kinase domain mutation analysis
Until date, more than 90 discrete resistance conferring point mutations at 57 residues in the ABL kinase have been documented.[7] However, many of these mutations are quite rare in imatinib treated clinical samples since 7 mutated codons (G250, Y253, E255, T315, M351, F359 and H396) together account for 60-70% of the total mutations.[27] As shown in Figure 1, these mutations generally fall within four regions of the KD: The ATP-binding P-loop (amino acids 248-256), the drug binding site (amino acids 315-317), the catalytic site (amino acids 350-363) and the activation loop (amino acids 381-402). The clinical interpretation and significance of finding a particular BCR-ABL KD mutation can be complex. The relative degree of imatinib resistance, defined by in vitro drug inhibition of kinase activity tends to be mutant specific with some mutations conferring only low-level resistance that may respond to imatinib dose escalation (e.g., M351T) and others conferring high-level resistance to imatinib and other TKIs (e.g., T315I, G250E, E255K), thus implying imatinib “failure” and the need for a change in therapy.[33]
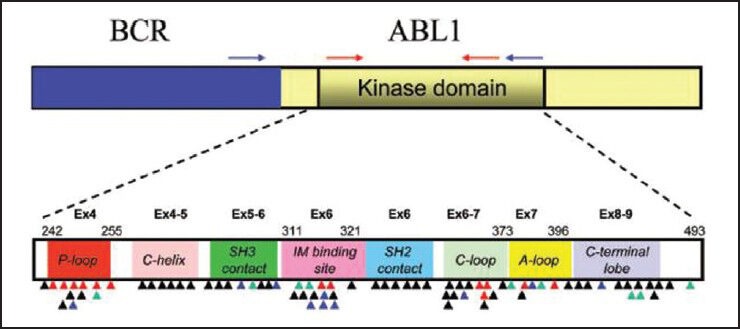
| Fig. 1 Schematic representation of the BCR-ABL transcript and the location of reported kinase domain (KD) mutations (The location of primers used for nested real-time polymerase chain reaction are indicated on the BCR and ABL genes. The external primers (blue arrow) are placed on BCR and ABL region of the fusion transcript. The internal primers (red arrows) are used to further amplify KD region of the fusion transcript. Colored triangles indicate the location of KD mutations reported in tyrosine kinase inhibitor (TKI)-resistant samples (black for imatinib, green for nilotinib/imatinib, blue for dasatinib/imatinib and red for all three TKIs). The KD subdomains, exons and amino acid numbers are shown. P-loop: Phosphate binding loop; IM binding site: Imatinib binding region; C-loop: Kinase catalytic domain; and A-loop: Activation loop [Adapted from Jones et al. (2009) J Mol Diag Vol.11, No. 1: 4-11])
Apart from imatinib, specific mutation types are also closely associated with resistance to 2nd generation TKIs and this information is useful in directing the choice of TKI after imatinib failure [Table 4]. Resistance to dasatinib often manifests as mutations at amino acids 299 (V299 L), 315 (T315I) and 317 (F317 L/I) and that to nilotinib preferentially results in mutations in the P-loop (G250E, Y253H, E255K), T315I, or F311I.[34] It appears that the spectrum of resistance mutations seen following use of these more powerful TKIs are more restricted than those seen following imatinib treatment, but often have complex dynamics dependent on the specific treatment regimen and the prior therapy. Common scenarios include (1) complete or incomplete clonal replacement of an imatinib-selected mutation with a completely different dasatinib or nilotinib selected clone; (2) new emergence of a BCR-ABL KD mutation only after exposure to a second-generation agent and (3) persistence of an imatinib-selected mutation plus the acquisition of an additional mutation after dasatinib/nilotinib exposure.[35]
Table 4
Selection of second generation tyrosine kinase inhibitor based on the mutation identified

Spectrum of mutations seen in Indian CML patients with resistance to imatinib
Between January 2007 and January 2010, peripheral blood and/or bone marrow samples from 1110 CML patients were received from various hospitals in India and were analyzed at our center for the presence of ABL KD mutations. To the best of our knowledge and as per the limited clinical information available with us, these samples pertained to CML patients in CP and on imatinib treatment. Though care has been taken to exclude patients of AP and BC from this analysis the possibility that some patients of AP might be a part of this cohort cannot be ruled out. In each case, the investigation for the presence of mutations was initiated by the treating Oncologist upon observation of clinical resistance in the patient while on imatinib.
Sensitivity and reliability of mutation detection is critically dependent on the quality and integrity of the sample RNA.[8] Total cellular RNA was extracted from leukocytes and reverse transcribed as previously reported.[9] All samples were first assessed for the level of BCR-ABL and ABL transcripts[36] and were subjected to mutation screening only if the RNA obtained from the sample contained a measurable level of BCR-ABL transcript and if the ABL control gene level indicated a non-degraded RNA. A nested real-time PCR for BCR-ABL transcript was performed such that the complete ABL KD was obtained as the final amplicon. Direct bidirectional sequencing of the amplicon was done on an ABI 3130xl Sequencer (Applied Biosystems, Foster City, CA) as per standard chemistries. The sequences were compared against the human genome sequence (NCBI accession NM_0073013) using BLAST software to identify the mutations. The chromatograms were also analyzed manually using SeqScape v2.6 software. The sensitivity of the method was 15-20% (data not shown).
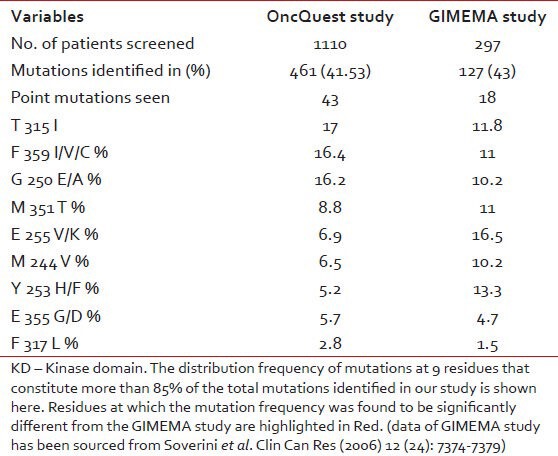
Bidirectional sequencing analysis showed the presence of one or more mutations in 461 of 1110 patients (41.53%). 23 patients exhibited more than one mutation simultaneously. A total of 43 discrete mutations mapped to 35 codons were detected. More than 85% of the total mutations were seen at one of the 9 residues: T315, F359, G250, M351, E255, M244, Y253, E355 and F317. The distribution and relative frequency of the mutations [Figure 2] were compared with those reported in a study carried out on the Caucasian race by the GIMEMA Working Party on CML and reported by Soverini et al. in 2006.[37] Significant differences were observed in the frequency of mutation at residues T315, F359, G250, E255, M244 and Y253. We observed a higher frequency of mutations at amino acids T315, F359 and G250 and a significantly lower frequency at residues M244, E255 and Y253 when compared with the study cited above (details are provided in [Table 5]. Overall, we found 29.2% of the total mutations falling in the P-loop region (codons 248-256) while a considerably higher percentage (46%of the total mutations) was reported in the GIMEMA study. It has been reported[8] that mutations are more frequently observed in the P-loop region in an advanced stage or progressive patients of CML. The lower frequency of P-loop mutations in our study may be attributed to the fact that while our study primarily focused on CP patients, the GIMEMA study had patients from all stages of CML (CP, AP, BC). 23 (4.9% of all mutated patients) patients in our study exhibited more than one mutation. Upon studying the domain localization of the mutations we noticed a trend towards preferential association of P-loop and drug binding domain mutations [Figure 3]. The specifics of this association in terms of disease stage and the temporal occurrence of the mutations will be analyzed further in a retrospective study on the retained samples of the patients.
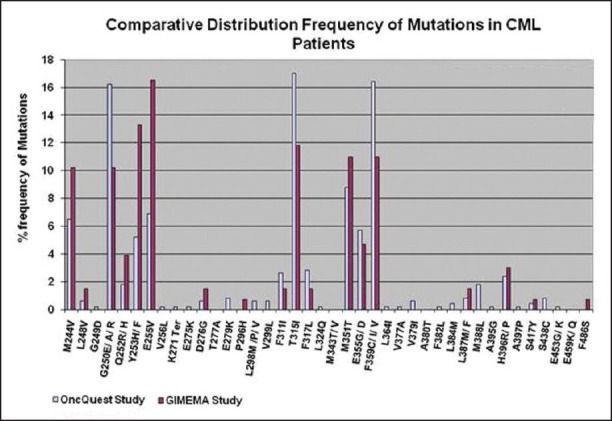
| Fig. 2 Distribution frequency of kinase domain (KD) mutations identified in Indian chronic myeloid leukemia (CML) patients (Bidirectional sequencing analysis of the BCR-ABL KD showed the presence of one or more mutations in 461 of 1110 patients (41.53%). A total of 43 discrete mutations mapped to 35 codons were detected. More than 85% of the total mutations were seen at one of the 9 residues: T315, F359, G250, M351, E255, M244, Y253, E355 and F317. The distribution and relative frequency of the mutations were compared to those reported in a study carried out on the Caucasian race by the GIMEMA Working Party on CML. Significant differences were observed in the frequency of mutation at residues T315, F359, G250, E255, M244 and Y253. A higher frequency of mutations at amino acids T315, F359 and G250 and a significantly lower frequency at residues M244, E255 and Y253 were observed in our study as compared to the study cited above [data of GIMEMA study has been sourced from Soverini et al. Clin Can Res (2006) 12 (24): 7374-7379])
Table 5
Comparative analysis of frequency of KD mutations identified in OncQuest and GIMEMA studies
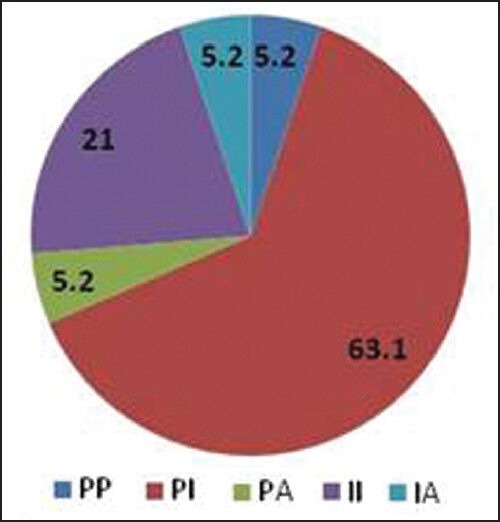
| Fig. 3 Domain-wise distribution of double mutants (4.9% of all mutated patients in our study exhibited more than one mutation. Upon studying the domain localization of the double mutants a significant association of P-loop and drug binding domain mutations was observed)
The aim of the present study was to investigate the presence of ABL KD mutations in Indian CML patients who exhibit imatinib resistance. This was done to assess the extent to which mutations account for or contribute to resistance in this group of patients. In our study, we found evidence of mutation in 41.32% patients. Such an incidence is somewhat lower than expected, but this could be mainly attributed to the fact that our study primarily focused on patients in CP of CML. It has been shown previously that the contribution of KD mutations to the resistant phenoltype was much lower in CP patients than in AP and BC patients, with lymphoid BC and Ph+ all patients having the greatest likelihood of harboring one, or even multiple, mutations.[37] Data from the Soverini study also showed that among CP patients mutation incidence in those who had received imatinib after a-IFN failure was approximately twice as high as in those who had received imatinib as first-line therapy (31%vs.14%). Such a significant difference between early and late CP supports the hypothesis that mutations tend to accumulate during the natural course of the disease as a result of a progressively increasing genetic instability. This observation finds further support from the report that KD mutations may be detected in a substantial fraction of imatinib naοve patients with advanced-phase CML.[20]
CONCLUSIONS
The clinical availability of imatinib has changed the natural history of CML, improving outcomes for a significant number of patients. However, imatinib does not benefit everyone and many patients may become resistance to it while on treatment. Increased understanding of the dynamics of leukemic response to TKI therapy has led to definitions of molecular responses that are optimal and those that are suboptimal. Suboptimal responses are associated with a significantly higher risk of mutations and loss of response. Regular molecular monitoring will allow loss of response to be recognized at an early stage in most cases and may also facilitate better compliance. Although dose escalation with imatinib has beneficial effects in some patients who experience imatinib resistance, the multiple mechanisms of resistance to imatinib highlight the requirement for alternate therapies. The resistance profile of each TKI to specific mutations has been better defined so that clinical recommendations based on these findings can now be established in terms of the 2nd generation TKIs to be used in the event of a specific mutation. The T315I mutation currently represents an important gap in the spectrum of drugs available to counter resistance and neither imatinib nor the second generation TKIs are effective against it. However, efforts are ongoing for the development of other second generation TKIs such as SKI-606, INNO-406 and LBH589 to ensure continued evolution of this therapeutic area.
ACKNOWLEDGMENTS
On behalf of OncQuest Laboratories Ltd., we gratefully acknowledge the support shown by the prescribing Oncologists in entrusting us with the monitoring and mutation analysis studies of their CML patients.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Schematic representation of the BCR-ABL transcript and the location of reported kinase domain (KD) mutations (The location of primers used for nested real-time polymerase chain reaction are indicated on the BCR and ABL genes. The external primers (blue arrow) are placed on BCR and ABL region of the fusion transcript. The internal primers (red arrows) are used to further amplify KD region of the fusion transcript. Colored triangles indicate the location of KD mutations reported in tyrosine kinase inhibitor (TKI)-resistant samples (black for imatinib, green for nilotinib/imatinib, blue for dasatinib/imatinib and red for all three TKIs). The KD subdomains, exons and amino acid numbers are shown. P-loop: Phosphate binding loop; IM binding site: Imatinib binding region; C-loop: Kinase catalytic domain; and A-loop: Activation loop [Adapted from Jones et al. (2009) J Mol Diag Vol.11, No. 1: 4-11])
| Fig. 2 Distribution frequency of kinase domain (KD) mutations identified in Indian chronic myeloid leukemia (CML) patients (Bidirectional sequencing analysis of the BCR-ABL KD showed the presence of one or more mutations in 461 of 1110 patients (41.53%). A total of 43 discrete mutations mapped to 35 codons were detected. More than 85% of the total mutations were seen at one of the 9 residues: T315, F359, G250, M351, E255, M244, Y253, E355 and F317. The distribution and relative frequency of the mutations were compared to those reported in a study carried out on the Caucasian race by the GIMEMA Working Party on CML. Significant differences were observed in the frequency of mutation at residues T315, F359, G250, E255, M244 and Y253. A higher frequency of mutations at amino acids T315, F359 and G250 and a significantly lower frequency at residues M244, E255 and Y253 were observed in our study as compared to the study cited above [data of GIMEMA study has been sourced from Soverini et al. Clin Can Res (2006) 12 (24): 7374-7379])
| Fig. 3 Domain-wise distribution of double mutants (4.9% of all mutated patients in our study exhibited more than one mutation. Upon studying the domain localization of the double mutants a significant association of P-loop and drug binding domain mutations was observed)