 PDF
PDF  Views
Views  Share
Share


Imaging Recommendations for Diagnosis, Staging, and Management of Pediatric Solid Tumors
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(01): 138-148
DOI: DOI: 10.1055/s-0042-1759507
Abstract
Paediatric extra-cranial solid tumours are one of the common causes for paediatric malignancies. Lack of appropriate imaging at presentation, staging and for follow-up is a major challenge for paediatric solid tumours. We have reviewed the paediatric solid tumour imaging protocols suggested by the major oncological societies/groups around the world (mainly the SIOP – Society International Pediatric Oncology, and the COG – Children's Oncology Group). We have adapted some of those protocols to develop imaging recommendations for the diagnosis, staging and management of extra-cranial solid tumours based on the treatment protocols followed in India.
Keywords
hepatoblastoma - imaging guidelines - neuroblastoma - pediatric oncology - pediatric solid tumors - Wilm's tumorNote
The article is not under consideration for publication elsewhere. Each author participated sufficiently for the work to be submitted. Publication is approved by all authors.
Publication History
Article published online:
06 March 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Paediatric extra-cranial solid tumours are one of the common causes for paediatric malignancies. Lack of appropriate imaging at presentation, staging and for follow-up is a major challenge for paediatric solid tumours. We have reviewed the paediatric solid tumour imaging protocols suggested by the major oncological societies/groups around the world (mainly the SIOP – Society International Pediatric Oncology, and the COG – Children's Oncology Group). We have adapted some of those protocols to develop imaging recommendations for the diagnosis, staging and management of extra-cranial solid tumours based on the treatment protocols followed in India.
Keywords
hepatoblastoma - imaging guidelines - neuroblastoma - pediatric oncology - pediatric solid tumors - Wilm's tumorChildhood cancer accounts for nearly 1%-of all cancers diagnosed worldwide across all age groups.[1] [2] Great improvement has been made in the last few decades in the treatment of childhood cancers, achieving successful treatment in up to 80%-of cases.[1] This dramatic success is a result of decades of collaborative effort by various study groups across the globe. Collaborative consensus guidelines have been developed by these groups for imaging and management of pediatric tumors.
This article aims to provide imaging guidelines for the common pediatric extracranial solid tumors based on the recommendations of various pediatric groups across the world and treatment protocols followed in India.
Clinical Presentation
Pediatric tumors present with nonspecific symptoms and requires high index of suspicion to investigate for the possibility of cancer. Pediatric abdominal malignancies present most commonly as abdominal distension or a palpable abdominal mass. It may be an incidentally detected mass or present with abdominal pain, hematuria, features of bowel or bladder obstruction, or may present with constitutional symptoms such as fatigue, loss of appetite or fever. Renal and suprarenal tumors may present with hypertension or paraneoplastic symptoms such as opsoclonus-myoclonus.[3] [4] Occasionally, they may present with precocious puberty/virilization, more so in adreno-cortical carcinomas, and rarely in hepatoblastoma and germ cell tumors.[5]
Soft tissue tumors may present with swelling or bone pain. Spinal extension of the tumor like in neuroblastoma or germ cell tumors may cause focal neurological deficits or bowel/bladder related symptoms.
Imaging Guidelines for Pediatric Abdominal Masses
Ultrasound
Ultrasound is the ideal screening tool in cases of suspected abdominal mass in children as there is no ionizing radiation or sedation involved.
Role of ultrasound is to confirm the presence of mass and determine its organ of origin. Further investigations would be based on the ultrasound findings ([Fig. 1]).
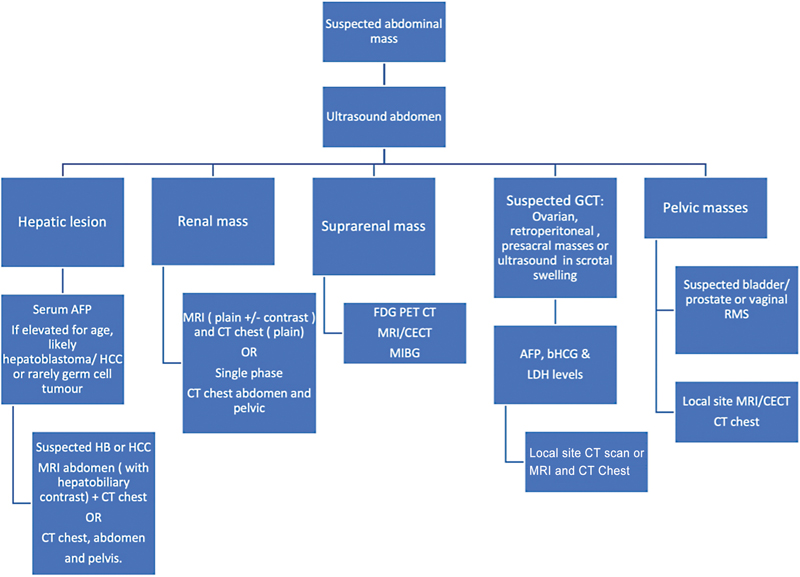
| Figure 1:Diagnostic pathways based on ultrasound findings.
Relook ultrasound after cross sectional study, along with Doppler might be useful to evaluate subtle vascular invasion or thrombosis in cases of doubt, e.g., tumor thrombus in hepatoblastoma or Wilm's tumor. It is a problem solving tool in cases of complex findings on cross-sectional imaging, e.g., suspected focal lesions in hepatoblastomas and lesions in contralateral kidney in cases of Wilm's tumor. In cases of suspected tumor rupture, ultrasound has a role in evaluating the tumor margin integrity and detecting echoes in the abdominal free fluid. USG-guided biopsy of solid pediatric tumors is a widely acceptable technique and again avoids unnecessary radiation (compared with CT scan).
Magnetic Resonance Imaging
MRI is the modality of choice for pediatric abdominal masses.[6] It provides better soft tissue contrast and does not expose to any ionizing radiation. However, sedation risk should be considered against radiation risk and CT scan can be performed for cases with contraindication to MRI or difficult anesthesia or no availability.[7] MRI field strength of 1.5 to 3 T and acquisition with the smallest suitable coil (e.g., head coil or flexible phased-array body coil) is recommended with breath holding and gated sequences. Study may be performed under sedation or general anesthesia if needed, depending on the age and weight of child.
CT Scan
A contrast-enhanced CT scan for hepatic or renal masses is the alternative imaging modality to MRI as per institute preference, or if MRI is contraindicated or anesthesia is not available. Care should be taken to use the lowest possible radiation dose with adequate image quality. Single-phase imaging is recommended if sufficient to provide necessary information. Just a non-contrast CT scan is usually avoided. The imaging protocols are further tailored as per the organ of origin of the mass and will be discussed subsequently.
A CT chest is a mandatory diagnostic procedure for all patients of liver and renal masses and protocol is described in the respective sections.
Pediatric Liver Tumors
Pediatric liver tumors are rare and ∼two-thirds of them are malignant, hepatoblastoma being the most common, (∼37%) followed by hepatocellular carcinoma (HCC; 21%), and sarcoma (8%).[8] Benign tumors such as hemangioma, hemangioendothelioma, focal nodular hyperplasia, and mesenchymal hamartomas form the rest of the spectrum.
Risk Factors, Etiopathogenesis, and Clinical Presentation
Hepatoblastoma are mostly sporadic, but may be associated with genetic abnormalities and familial cancer syndromes, such as the Beckwith–Wiedemann syndrome and familial adenomatous polyposis.[9] Premature birth and very low birth weight are known to be associated with increased incidence of hepatoblastoma.[10]
The most common presenting symptom is abdominal distension or a palpable abdominal mass. It may be associated with non-specific symptoms such as abdominal discomfort, fatigue, and loss of appetite, and the child may appear pale due to anemia, especially in HCC and liver sarcomas
Imaging Guidelines for Pediatric Liver Lesions
Ultrasonography
USG is the ideal screening modality for suspected liver mass/abdominal lump.[6] [11]
Contrast-enhanced USG can be performed for the initial assessment of lesion to help classify as benign or malignant. USG may be useful to evaluate hepatic veins, IVC, portal vein, and focal liver lesions and for suspected cases of tumor rupture.
Magnetic Resonance Imaging
Both MRI and CECT are the modality of choice for pediatric liver masses such as hepatoblastoma and HCC.
Respiratory gated sequences should be used. Unlike adults, breath holding is usually not possible for hepatoblastoma evaluation as these scans are generally performed under sedation or GA. The recommended MRI sequences are presented in [Table 1].
|
MRI sequence |
Plane |
Thickness |
Sequence coverage |
|---|---|---|---|
|
T2 STIR |
Coronal |
5 mm |
Above diaphragm to below iliac crests |
|
T1 TSE FS |
Coronal |
2 mm |
Above diaphragm to below iliac crests |
|
T1 TSE FS |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T2W volume sequence (SPACE/VISTA) |
Coronal |
0.9 mm–1.1 mm |
Above diaphragm to below iliac crests |
|
T2W STIR |
Axial |
5 mm |
To include area of interest |
|
T1W IP and OP |
Axial |
5 mm |
To include area of interest |
|
DWI/ADC (b = 0,100,500,1000) |
Axial |
6 mm |
To include area of interest |
|
Post-contrast |
|||
|
T1W FS immediate (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 30 s arterial (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 60 s venous (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 2 min (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 5 min (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 10 min (VIBE) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
T1W TSE FS |
Axial |
5 mm |
Above diaphragm to below iliac crests |
|
T1W FS 20 min (VIBE) |
Coronal |
2 mm |
Above diaphragm to below iliac crests |
|
T1W FS 25 min (vibe) |
Axial |
2 mm |
Above diaphragm to below iliac crests |
|
Imaging recommendation |
Protocol |
Contrast dose |
Reconstructions |
|---|---|---|---|
|
CT Abdomen ± Chest |
-To include lung apices to lesser trochanter -Inject two-thirds of contrast bolus @ 0.5 mL per sec and one-third @ 1 mL per second -Scan after 10 seconds post injection Slice thickness – 0.6 mm Increment – 0.6 mm Pitch – 0 |
2 mL/kg (dose recommendations for Iohexol 300)* |
Lung window: - Axial 1 mm - Cor 3 mm - -Sag 3 mm Soft tissue window (chest and abdomen): - Axial 1 mm - Cor 3 mm - Sag 3 mm Bone window: - Cor 2 mm - Sag 2 mm |
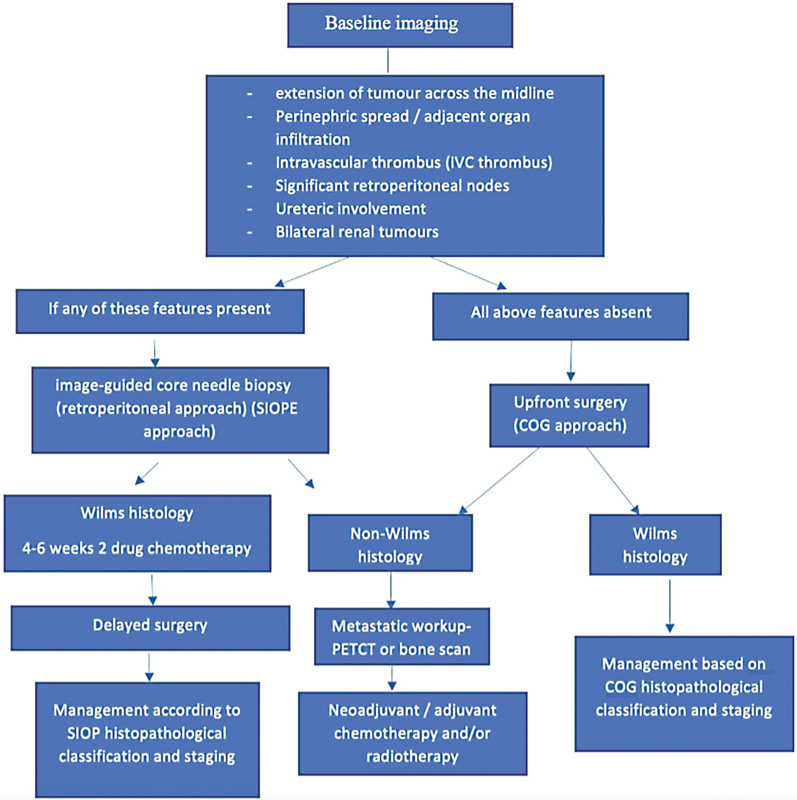
| Figure 2:Guidelines for the imaging and management of pediatric renal tumors.
Radical nephroureterectomy and lymph node sampling is the surgical procedure of choice[17] Clearance of the thrombus, if present, is performed and either cavotomy or partial cavectomy may be required depending on the extent of tumor thrombus.[18] As Wilm's tumor is highly chemo-sensitive, post-operative chemotherapy is recommended for every patient, and the dosage regimen is decided based on the histology and tumor stage ([Supplementary Table S2], available online only). Except for stage 1 and 2 tumors with favorable histology, post-operative radiotherapy is indicated for primary tumor bed and metastatic disease.[16] [19]
Upfront surgery is contraindicated in bilateral renal disease and in cases with solitary or horseshoe kidney.[16] [20] For patients with bilateral Wilm's tumors, neoadjuvant chemotherapy is initiated, followed by delayed surgery in the form of bilateral partial nephrectomy or total nephrectomy on the worse side and partial nephrectomy on the opposite side. Patients with tumor spillage/rupture, or regional lymph node metastasis are upstaged to stage III, and should receive abdominal radiotherapy and appropriate chemotherapy.[19] [21]
Follow-up of patients: Done with USG of the abdomen and pelvis along with chest radiograph ([Supplementary Table S2], available online only).[11]
Extracranial Germ Cell Tumors (Pediatric)
Malignant germ cell tumors (GCT) account for 3%-of pediatric cancer with bimodal age distribution.[22] [23] Sites are divided as intracranial or extracranial, the later as gonadal and extragonadal. The extragonadal sites comprise sacrococcygeal, mediastinal, retroperitoneal, and other para-axial locations. Histologically, the germ cell tumors are classified as germinoma (dysgerminoma and seminoma), and non-germinomatous tumors such as endodermal sinus (yolk sac tumor), embryonal carcinoma and choriocarcinoma, or mixed type where more than one histology co-exist.
Risk Factors, Etiopathogenesis, and Clinical Presentation
Cryptorchidism and gonadal dysgenesis are associated with an increased risk of the development of gonadal GCT.[24] Some GCTs are more frequently seen with sex-linked chromosomal disorders; mediastinal GCT in Klinefelter's syndrome, germinomas in Turner's syndrome, and Swyer's syndrome.
Clinical features are variable and the presentation depends on the site of the extracranial GCT.[24] Sacrococcygeal GCT usually present as an external palpable mass in perinatal period. Testicular GCT may present as a painless swelling. Ovarian and abdominal or retroperitoneal GCT present with gradual abdominal distension and discomfort. Rarely ovarian GCT may present as acute abdomen due to torsion, rupture, or intralesional hemorrhage. Mediastinal GCT causes symptoms due to mass effects such as airway compression, superior vena cava obstruction, or heart failure.
Diagnostic Work-up of Germ Cell Tumors
Tumor markers important for the diagnosis of GCT are α fetoprotein (aFP) secreted by yolk sac tumors and β human chorionic gonadotropin (b-hCG) secreted by choriocarcinoma. LDH is a non-specific marker of tumor burden.[25] The imaging recommended at baseline and for follow-up is summarized in [Supplementary Table S3], available online only.
USG
USG is the modality of choice for the evaluation of suspected testicular pathology. Trans-scrotal USG using a linear high-frequency probe has more than 90%- sensitivity and specificity in detection of testicular malignancies. In adjunct to gray scale and color Doppler, elastography provides an added value.[26] USG is usually the initial imaging modality for abdominal and pelvic GCT.
Cross-sectional Imaging
Primary site evaluation can be performed with CT scan or MRI based on the site involved and local institutional policy.
CT Scan
Contrast-enhanced single venous phase CT scan of the chest, abdomen, and pelvis is suggested for initial staging of abdominal GCT.[25] [27] It is a sensitive modality for assessing retroperitoneal nodes. Noncontrast CT scan of the chest is recommended for the evaluation of lung metastasis
MRI Scan
MRI is used as an adjunct tool to USG, in cases of testicular mass with diagnostic dilemma on ultrasound. MRI abdomen and pelvis may be used instead of CT for both baseline evaluation and post treatment reassessment. MRI may particularly be used for initial workup in patients with deranged renal profile or in cases with suspicious intraspinal extension.
Contrast CT or MRI of the head should be performed if brain metastasis is suspected and in all patients with metastatic choriocarcinoma.
Recommendations for Biopsy: Biopsy is performed if AFP is not elevated and neoadjuvant chemotherapy is planned. Biopsy is not recommended for upfront resection or in testicular tumors where high orchidectomy is planned. Biopsy is performed when tumor markers are normal and/or there is a clinicoradiological mismatch.[27]
Management
A multi-modality customized strategy is needed for the treatment of GCT depending on the site, stage, and tumor biology. The COG staging for testicular, ovarian, and extragonadal germ cell tumors is recommended. GCTs are stratified into low-, intermediate-, and high-risk categories.
Surgery is the mainstay of management and first treatment option if feasible. A biopsy with neoadjuvant chemotherapy is otherwise recommended.
Neuroblastoma
Neuroblastoma (NB) is a neuroectodermal tumor arising from the sympathetic ganglion cells and is the commonest extracranial solid tumor in children.[28] This tumor commonly arises in the adrenal gland and can also occur at multiple locations including the extraadrenal abdomen, thorax, neck, and pelvis along the course of sympathetic nervous system.[29] Patients usually present with symptoms caused due to its mass effect on the surrounding organs, mostly presenting as a large abdominal mass.
Imaging Guidelines and Principles of Management
The treatment and outcome of NB are dependent on risk assessment and stage of the disease. The international Neuroblastoma Risk Group (INRG) task force puts forth a preoperative staging called International Neuroblastoma Risk Group Staging System (INRGSS), which is dependent on cross-sectional imaging (CECT or MRI) of the tumor site using Imaging defined risk factors (IDRF) ([Supplementary Tables S4] and [S5], available online only). A metastatic workup includes an iodine-labeled MIBG scan (radionuclide 123/124 or 131 iodine is used; however, in India, only 131 iodine is available) to look for sites of metastatic disease, especially bone lesions, a bone marrow biopsy, and aspirate for marrow disease.[29] [30] The IDRFs on cross-sectional imaging help categorize the patients into L1 (non-infiltrative/operable tumor) or L2 (infiltrative or disease-encasing important structures) ([Supplementary Table S6], available online only).[29] Combining the outcome of these tests with other pathological and molecular criteria, and age of the patient help make decisions for management plan.
Data are emerging for the potential role of FDG PET-CT in the evaluation of neuroblastoma. In the Indian set-up, due to the unavailability of I-123 for MIBG scans, I-131 MIBG is performed. For the MIBG non-avid NB lesions, PET-CT is preferred modality for metastatic work-up. The utility of PET-CT for all NB patients work-up is debatable as there are some studies that have shown higher sensitivity of PET-CT compared with I-123 MIBG scans for stage 1 or 2 disease, but MIBG has performed better for higher stages of NB.[31]
Patients are treated with surgery alone in L1, low-risk disease or initially with chemotherapy followed by surgery, which could be a complete resection or an incomplete resection in patients with intermediate risk disease (patients having an unfavorable histology or infantile age group with metastasis receiving additional cycles of chemotherapy). Patients with high-risk neuroblastoma undergo bone marrow transplant after initial intensive chemotherapy and surgery followed by radiotherapy and administration of differentiation agents and immunotherapy if available.
MRI/CT and MIBG scan along with bone marrow biopsy are done pre-surgically to look for residual disease, as this may alter the management of the disease.
Rhabdomyosarcoma
Introduction
Rhabdomyosarcoma (RMS) is the most common childhood soft tissue sarcoma and accounts for 3 to 5%-of all pediatric malignancies. It is the third most common soft tissue tumor in children after neuroblastoma and Wilm's tumor.[32] RMS in children has two main histological subtypes, embryonal and alveolar. Further refinement of prognosis has occurred over the years with the incorporation of molecular fusion status for PAX 3/7-FOXO1 with fusion-positive tumors faring worse than fusion-negative ones. The embryonal type (nearly 75%-of RMS cases), which is mostly fusion negative, is more common in younger children (<10>
Risk Factors and Etiopathogenesis and Clinical Presentation
There are no clear risk factors for RMS. Higher risk of developing RMS has been shown in children who have the following rare, inherited conditions: Li-Fraumeni syndrome, Beckwith–Wiedemann syndrome, neurofibromatosis, DICER 1, cardio-facio-cutaneous syndrome, Costello syndrome.[33]
There are no clear signs or symptoms for RMS and the presentation is largely dependent on the location of the lesion. RMS is known to have metastasis at presentation in 15 to 20%-of cases, and the common sites for metastasis are lungs, bone marrow, bones, and distant lymph nodes.[34] Tumor biopsy is routinely performed as part of the main tumor work-up and at least one bone marrow aspirate and trephine performed for evaluating bone marrow involvement. The metastatic work-up includes lymph node biopsy, bilateral bone marrow aspirates, and CSF examination when LN, bone marrow, or neurological metastasis is suspected.
The RMS cases are risk stratified into low-risk, intermediate-risk, high-risk or very high-risk category based on age, tumor size, regional nodal status, tumor site, histology, PAX-FOX01 status, and Intergroup Rhabdomyosarcoma Study (IRS) post-surgical stage. Treatment for RMS cases is multimodality and in low/intermediate risk categories, the approach is chemotherapy in combination with radiotherapy and/or surgery with additional maintenance chemotherapy in high risk cases. Localized radiotherapy for metastatic disease can be used. In spite of recent advancements, multicenter trials and significantly improved treatment protocols, the 5-year overall survival for pediatric RMS stands at 75%-for cases with localized disease, dropping to just 30%-for cases with metastatic disease.[35] [36]
Imaging Guidelines
Imaging the primary tumor should include loco-regional lymph node sites and also cover the regional extent of the tumor including the neuro-vascular structures. Evaluation for metastasis should include chest CT scan and bone scan or PET-CT for complete staging. Complete imaging and staging should be performed before biopsy is performed. All the imaging should be planned and reported by a pediatric radiologist with oncology experience and a nuclear medicine physician for hybrid imaging. Imaging reports should clearly describe the lesion location, size, lesion characteristics, extent and status of surrounding structures including regional lymph nodes.
MRI
MRI is the imaging modality of choice for both initial imaging of the tumor and subsequent follow-up examinations ([Table 3]). The MRI protocol should include DWI/ADC and post-contrast imaging. Lesions with diffusion restriction and lower ADC values have shown to correlate with poor outcome and higher incidence of recurrence.[37] Post-contrast tumor enhancement helps to map tumor response and also study neurovascular spread (for head and neck RMS). The MRI field of view should include areas of loco-regional lymph nodes.
|
MRI sequence |
Plane |
Thickness |
Sequence coverage |
|---|---|---|---|
|
T2 STIR |
Sag |
5 mm |
Pelvic lesion only (below liver to symphysis pubis) |
|
T1 TSE |
Axial |
5 mm |
Above diaphragm to below symphysis pubis |
|
T1 TSE FS |
Axial |
2 mm |
Above diaphragm to below symphysis pubis |
|
T2W volume sequence (SPACE/VISTA) |
Coronal |
0.9 mm–1.1 mm |
Above diaphragm to below symphysis pubis |
|
T2W STIR |
Axial |
5 mm |
To include area of interest |
|
DWI/ADC (b = 0,100,500,1000) |
Axial |
6 mm |
To include area of interest |
|
Post-contrast |
|||
|
T1W FS (VIBE) 1 |
Axial |
2 mm |
Above diaphragm to below symphysis pubis |
|
T1W FS (VIBE) |
Coronal/Sag |
2 mm |
Above diaphragm to below symphysis pubis |
|
T1W FS (VIBE) 2 |
Axial |
2 mm |
Above diaphragm to below symphysis pubis |
|
MRI sequence |
Plane |
Thickness |
Sequence coverage |
|---|---|---|---|
|
Essential sequences |
|||
|
T2 W FSE/TSE (TE ≥ 120 ms) Fat-saturated |
Axial |
≤ 2 mm |
Both orbits |
|
T1 W TSE/FSE |
Axial |
≤ 2 mm |
Both orbits |
|
T2W TSE/FSE |
Sagittal oblique |
≤ 2 mm |
Both orbits |
|
T2W fat-saturated/STIR |
Coronal |
≤ 2 mm |
Both orbits |
|
Post contrast (PC) |
|||
|
PC T1W SE FS/nonFS |
Axial |
≤ 2 mm |
Both orbits |
|
PC T1W SE FS/nonFS |
Coronal |
≤ 2 mm |
Both orbits |
|
PC T1W 2D or (3D GRE ≤ 1 mm) |
Axial |
≤ 3 mm |
Brain |
|
Optional sequences |
|||
|
3D T2W (CISS/SPACE/FIESTA/ DRIVE) |
Axial |
<1> |
Both orbits |
|
PC whole brain T1W 3D MPR |
Axial |
<1> |
|
|
PC whole Spine T1W |
Sagittal |
<3> |
Required if there is optic nerve meningeal sheath involvement |
References
- Bhakta N, Force LM, Allemani C. et al. Childhood cancer burden: a review of global estimates. Lancet Oncol 2019; 20 (01) e42-e53
- Johnston WT, Erdmann F, Newton R, Steliarova-Foucher E, Schüz J, Roman E. Childhood cancer: Estimating regional and global incidence. Cancer Epidemiol 2021; 71 (Pt B): 101662
- Kushner BH, Khakoo Y. Enigmatic entities: opsoclonus myoclonus ataxia syndrome linked to neuroblastoma. Lancet Child Adolesc Health 2018; 2 (01) 3-5
- Maas MH, Cransberg K, van Grotel M, Pieters R, van den Heuvel-Eibrink MM. Renin-induced hypertension in Wilms tumor patients. Pediatr Blood Cancer 2007; 48 (05) 500-503
- Yhoshu E, Lone YA, Mahajan JK, Singh UB. Hepatoblastoma with precocious puberty. J Indian Assoc Pediatr Surg 2019; 24 (01) 68-71
- Schooler GR, Squires JH, Alazraki A. et al. Pediatric hepatoblastoma, hepatocellular carcinoma, and other hepatic neoplasms: Consensus Imaging Recommendations from American College of Radiology Pediatric Liver Reporting and Data System (LI-RADS) Working Group. Radiology 2020; 296 (03) 493-497
- Callahan MJ, Cravero JP. Should I irradiate with computed tomography or sedate for magnetic resonance imaging?. Pediatr Radiol 2022; 52 (02) 340-344
- Ng K, Mogul DB. Pediatric liver tumors. Clin Liver Dis 2018; 22 (04) 753-772
- Czauderna P, Lopez-Terrada D, Hiyama E, Häberle B, Malogolowkin MH, Meyers RL. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 2014; 26 (01) 19-28
- Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer 2012; 59 (05) 776-779
- Towbin AJ, Meyers RL, Woodley H. et al. 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 2018; 48 (04) 536-554
- Agarwala S, Gupta A, Bansal D. et al. Management of hepatoblastoma: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 456-464
- Cunningham ME, Klug TD, Nuchtern JG. et al. Global disparities in Wilms tumor. J Surg Res 2020; 247: 34-51
- Charlton J, Irtan S, Bergeron C, Pritchard-Jones K. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 2017; 19: e8
- Nelson MV, van den Heuvel-Eibrink MM, Graf N, Dome JS. New approaches to risk stratification for Wilms tumor. Curr Opin Pediatr 2021; 33 (01) 40-48
- Qureshi SS, Kembhavi SA, Bhagat M. et al. Customized approach for upfront or delayed resection using radiological criteria in unilateral, nonmetastatic pediatric renal tumors: a prospective study. Pediatr Blood Cancer 2019; 66 (Suppl. 03) e27815
- Qureshi SS, Bhagat M, Kazi M. et al. Standardizing lymph nodal sampling for Wilms tumor: a feasibility study with outcomes. J Pediatr Surg 2020; 55 (12) 2668-2675
- Qureshi SS, Bhagat M, Smriti V. et al. Intravascular extension of Wilms tumor: Characteristics of tumor thrombus and their impact on outcomes. J Pediatr Urol 2021; 17 (01) 69.e1-69.e8
- NCG guidelines. 2020. Paediatric hematolymphoid and solid tumours. Accessed November 07, 2022, at: https://tmc.gov.in/ncg/index.php/107-guidlines
- Kembhavi SA, Qureshi S, Vora T. et al. Understanding the principles in management of Wilms' tumour: can imaging assist in patient selection?. Clin Radiol 2013; 68 (07) 646-653
- Prasad M, Vora T, Agarwala S. et al. Management of Wilms tumor: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 437-445
- Shaikh F, Murray MJ, Amatruda JF. et al. Paediatric extracranial germ-cell tumours. Lancet Oncol 2016; 17 (04) e149-e162
- Faure-Conter C, Rocourt N, Sudour-Bonnange H. et al. [Pediatric germ cell tumours]. Bull Cancer 2013; 100 (04) 381-391
- Horton Z, Schlatter M, Schultz S. Pediatric germ cell tumors. Surg Oncol 2007; 16 (03) 205-213
- Agarwala S, Mitra A, Bansal D. et al. Management of pediatric malignant germ cell tumors: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 465-472
- Dieckmann KP, Frey U, Lock G. Contemporary diagnostic work-up of testicular germ cell tumours. Nat Rev Urol 2013; 10 (12) 703-712
- Feldman DR. State-of-the-art management of germ cell tumors. Am Soc Clin Oncol Educ Book 2018; 38 (38) 319-323
- Matthay KK, Shulkin B, Ladenstein R. et al. Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neuroblastoma Risk Group (INRG) Task Force. Br J Cancer 2010; 102 (09) 1319-1326
- Monclair T, Brodeur GM, Ambros PF. et al; INRG Task Force. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 2009; 27 (02) 298-303
- Kembhavi SA, Shah S, Rangarajan V, Qureshi S, Popat P, Kurkure P. Imaging in neuroblastoma: An update. Indian J Radiol Imaging 2015; 25 (02) 129-136
- Sharp SE, Shulkin BL, Gelfand MJ, Salisbury S, Furman WL. 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 2009; 50 (08) 1237-1243
- Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014; 64 (02) 83-103
- Ripperger T, Bielack SS, Borkhardt A. et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A 2017; 173 (04) 1017-1037
- Skapek SX, Ferrari A, Gupta AA. et al. Rhabdomyosarcoma. Nat Rev Dis Primers 2019; 5 (01) 1
- Arndt CAS, Bisogno G, Koscielniak E. Fifty years of rhabdomyosarcoma studies on both sides of the pond and lessons learned. Cancer Treat Rev 2018; 68: 94-101
- Hibbitts E, Chi YY, Hawkins DS. et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: a report from the Children's Oncology Group. Cancer Med 2019; 8 (14) 6437-6448
- van Ewijk R, Schoot RA, Sparber-Sauer M. et al; Cooperative Weichteilsarkom Studiengruppe Imaging Group, the European Society of Paediatric Radiology Oncology Task Force and the European Paediatric Soft Tissue Sarcoma Study Group Imaging Committee. European guideline for imaging in paediatric and adolescent rhabdomyosarcoma - joint statement by the European Paediatric Soft Tissue Sarcoma Study Group, the Cooperative Weichteilsarkom Studiengruppe and the Oncology Task Force of the European Society of Paediatric Radiology. Pediatr Radiol 2021; 51 (10) 1940-1951
- O JH, Lodge MA, Wahl RL. O JH. Practical PERCIST: a simplified guide to PET response criteria in solid tumors 1.0. Radiology 2016; 280 (02) 576-584
- Accessed November 07, 2022, at: https://tmc.gov.in/tmh/pdf/EBM 2018-Guidelines for Paediatric Solid Tumours.pdf
- Crist WM, Anderson JR, Meza JL. et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 2001; 19 (12) 3091-3102
- Raney RB, Walterhouse DO, Meza JL. et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol 2011; 29 (10) 1312-1318
- Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 2009; 93 (09) 1129-1131
- Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am 2015; 62 (01) 201-223
- Rangamani S, SathishKumar K, Manoharan N. et al. Paediatric retinoblastoma in India: evidence from the National Cancer Registry Programme. Asian Pac J Cancer Prev 2015; 16 (10) 4193-4198
- Stathopoulos C,
Lumbroso-Le Rouic L, Moll AC. et al. Current indications of
secondary enucleation in retinoblastoma management: a position paper on behalf of the European Retinoblastoma
Group (EURbG). Cancers (Basel) 2021; 13 (14) 3392
Address for correspondence
Tejas Kapadia, DNB, FRCR, MMedChildren's X-ray Department/Academic Unit of Paediatric RadiologyOxford Road, M13 9WL ManchesterUnited KingdomEmail: tejaskap@gmail.comPublication History
Article published online:
06 March 2023© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1:Diagnostic pathways based on ultrasound findings.
| Figure 2:Guidelines for the imaging and management of pediatric renal tumors.
References
- Bhakta N, Force LM, Allemani C. et al. Childhood cancer burden: a review of global estimates. Lancet Oncol 2019; 20 (01) e42-e53
- Johnston WT, Erdmann F, Newton R, Steliarova-Foucher E, Schüz J, Roman E. Childhood cancer: Estimating regional and global incidence. Cancer Epidemiol 2021; 71 (Pt B): 101662
- Kushner BH, Khakoo Y. Enigmatic entities: opsoclonus myoclonus ataxia syndrome linked to neuroblastoma. Lancet Child Adolesc Health 2018; 2 (01) 3-5
- Maas MH, Cransberg K, van Grotel M, Pieters R, van den Heuvel-Eibrink MM. Renin-induced hypertension in Wilms tumor patients. Pediatr Blood Cancer 2007; 48 (05) 500-503
- Yhoshu E, Lone YA, Mahajan JK, Singh UB. Hepatoblastoma with precocious puberty. J Indian Assoc Pediatr Surg 2019; 24 (01) 68-71
- Schooler GR, Squires JH, Alazraki A. et al. Pediatric hepatoblastoma, hepatocellular carcinoma, and other hepatic neoplasms: Consensus Imaging Recommendations from American College of Radiology Pediatric Liver Reporting and Data System (LI-RADS) Working Group. Radiology 2020; 296 (03) 493-497
- Callahan MJ, Cravero JP. Should I irradiate with computed tomography or sedate for magnetic resonance imaging?. Pediatr Radiol 2022; 52 (02) 340-344
- Ng K, Mogul DB. Pediatric liver tumors. Clin Liver Dis 2018; 22 (04) 753-772
- Czauderna P, Lopez-Terrada D, Hiyama E, Häberle B, Malogolowkin MH, Meyers RL. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 2014; 26 (01) 19-28
- Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer 2012; 59 (05) 776-779
- Towbin AJ, Meyers RL, Woodley H. et al. 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 2018; 48 (04) 536-554
- Agarwala S, Gupta A, Bansal D. et al. Management of hepatoblastoma: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 456-464
- Cunningham ME, Klug TD, Nuchtern JG. et al. Global disparities in Wilms tumor. J Surg Res 2020; 247: 34-51
- Charlton J, Irtan S, Bergeron C, Pritchard-Jones K. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 2017; 19: e8
- Nelson MV, van den Heuvel-Eibrink MM, Graf N, Dome JS. New approaches to risk stratification for Wilms tumor. Curr Opin Pediatr 2021; 33 (01) 40-48
- Qureshi SS, Kembhavi SA, Bhagat M. et al. Customized approach for upfront or delayed resection using radiological criteria in unilateral, nonmetastatic pediatric renal tumors: a prospective study. Pediatr Blood Cancer 2019; 66 (Suppl. 03) e27815
- Qureshi SS, Bhagat M, Kazi M. et al. Standardizing lymph nodal sampling for Wilms tumor: a feasibility study with outcomes. J Pediatr Surg 2020; 55 (12) 2668-2675
- Qureshi SS, Bhagat M, Smriti V. et al. Intravascular extension of Wilms tumor: Characteristics of tumor thrombus and their impact on outcomes. J Pediatr Urol 2021; 17 (01) 69.e1-69.e8
- NCG guidelines. 2020. Paediatric hematolymphoid and solid tumours. Accessed November 07, 2022, at: https://tmc.gov.in/ncg/index.php/107-guidlines
- Kembhavi SA, Qureshi S, Vora T. et al. Understanding the principles in management of Wilms' tumour: can imaging assist in patient selection?. Clin Radiol 2013; 68 (07) 646-653
- Prasad M, Vora T, Agarwala S. et al. Management of Wilms tumor: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 437-445
- Shaikh F, Murray MJ, Amatruda JF. et al. Paediatric extracranial germ-cell tumours. Lancet Oncol 2016; 17 (04) e149-e162
- Faure-Conter C, Rocourt N, Sudour-Bonnange H. et al. [Pediatric germ cell tumours]. Bull Cancer 2013; 100 (04) 381-391
- Horton Z, Schlatter M, Schultz S. Pediatric germ cell tumors. Surg Oncol 2007; 16 (03) 205-213
- Agarwala S, Mitra A, Bansal D. et al. Management of pediatric malignant germ cell tumors: ICMR consensus document. Indian J Pediatr 2017; 84 (06) 465-472
- Dieckmann KP, Frey U, Lock G. Contemporary diagnostic work-up of testicular germ cell tumours. Nat Rev Urol 2013; 10 (12) 703-712
- Feldman DR. State-of-the-art management of germ cell tumors. Am Soc Clin Oncol Educ Book 2018; 38 (38) 319-323
- Matthay KK, Shulkin B, Ladenstein R. et al. Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neuroblastoma Risk Group (INRG) Task Force. Br J Cancer 2010; 102 (09) 1319-1326
- Monclair T, Brodeur GM, Ambros PF. et al; INRG Task Force. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 2009; 27 (02) 298-303
- Kembhavi SA, Shah S, Rangarajan V, Qureshi S, Popat P, Kurkure P. Imaging in neuroblastoma: An update. Indian J Radiol Imaging 2015; 25 (02) 129-136
- Sharp SE, Shulkin BL, Gelfand MJ, Salisbury S, Furman WL. 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 2009; 50 (08) 1237-1243
- Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014; 64 (02) 83-103
- Ripperger T, Bielack SS, Borkhardt A. et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A 2017; 173 (04) 1017-1037
- Skapek SX, Ferrari A, Gupta AA. et al. Rhabdomyosarcoma. Nat Rev Dis Primers 2019; 5 (01) 1
- Arndt CAS, Bisogno G, Koscielniak E. Fifty years of rhabdomyosarcoma studies on both sides of the pond and lessons learned. Cancer Treat Rev 2018; 68: 94-101
- Hibbitts E, Chi YY, Hawkins DS. et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: a report from the Children's Oncology Group. Cancer Med 2019; 8 (14) 6437-6448
- van Ewijk R, Schoot RA, Sparber-Sauer M. et al; Cooperative Weichteilsarkom Studiengruppe Imaging Group, the European Society of Paediatric Radiology Oncology Task Force and the European Paediatric Soft Tissue Sarcoma Study Group Imaging Committee. European guideline for imaging in paediatric and adolescent rhabdomyosarcoma - joint statement by the European Paediatric Soft Tissue Sarcoma Study Group, the Cooperative Weichteilsarkom Studiengruppe and the Oncology Task Force of the European Society of Paediatric Radiology. Pediatr Radiol 2021; 51 (10) 1940-1951
- O JH, Lodge MA, Wahl RL. O JH. Practical PERCIST: a simplified guide to PET response criteria in solid tumors 1.0. Radiology 2016; 280 (02) 576-584
- Accessed November 07, 2022, at: https://tmc.gov.in/tmh/pdf/EBM 2018-Guidelines for Paediatric Solid Tumours.pdf
- Crist WM, Anderson JR, Meza JL. et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 2001; 19 (12) 3091-3102
- Raney RB, Walterhouse DO, Meza JL. et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol 2011; 29 (10) 1312-1318
- Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 2009; 93 (09) 1129-1131
- Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am 2015; 62 (01) 201-223
- Rangamani S, SathishKumar K, Manoharan N. et al. Paediatric retinoblastoma in India: evidence from the National Cancer Registry Programme. Asian Pac J Cancer Prev 2015; 16 (10) 4193-4198
- Stathopoulos C, Lumbroso-Le Rouic L, Moll AC. et al. Current indications of secondary enucleation in retinoblastoma management: a position paper on behalf of the European Retinoblastoma Group (EURbG). Cancers (Basel) 2021; 13 (14) 3392