 PDF
PDF  Views
Views  Share
Share


Hepatic Amyloidosis: Something That Can camouflage and Deceive our Perception!
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(02): 236-239
DOI: DOI: 10.4103/ijmpo.ijmpo_46_16
Abstract
Amyloidosis is a multi-systemic diffusely infiltrating disease due to extracellular deposition of protein-mucopolysaccharide complexes. The type of protein deposited determines the subgroup of amyloid. Hepatic amyloidosis is a rare infiltrating disease affecting the hepatic parenchyma. A wide range of clinical presentation and atypical imaging findings delay the diagnosis of amyloidosis, while tissue biopsy demonstrating amyloid deposits is vital for a definitive diagnosis.
Keywords
B mode ultrasound - contrast CT - hepatic amyloidosis - infiltrating disease - primary secondary amyloidosisPublication History
Article published online:
06 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Amyloidosis is a multi-systemic diffusely infiltrating disease due to extracellular deposition of protein-mucopolysaccharide complexes. The type of protein deposited determines the subgroup of amyloid. Hepatic amyloidosis is a rare infiltrating disease affecting the hepatic parenchyma. A wide range of clinical presentation and atypical imaging findings delay the diagnosis of amyloidosis, while tissue biopsy demonstrating amyloid deposits is vital for a definitive diagnosis.
Introduction
Amyloidosis is a multi-systemic diffusely infiltrating disease due to extracellular deposition of fibrils of protein-mucoplysaccharide complexes. Hepatic amyloidosis is a rare infiltrating disease affecting the hepatic parenchyma. A wide range of clinical presentation and atypical imaging findings delay a diagnosis of amyloidosis. Tissue biopsy demonstrating amyloid deposits is vital for a definite diagnosis.
To get a detailed insight of hepatic amyloidosis, we present a case of primary amyloidosis of liver presented to our hospital with atypical clinical and imaging findings.
Case Report
A 56-year-old male patient presented at Gastro-medicine Department with a history of abdominal distension, anorexia, weight loss, pedal edema for the past 3 months, which are progressively increasing. Had consulted an outside hospital, outside ultrasound abdomen showed hepatosplenomegaly with ascites. He came to our hospital for further evaluation and management. Blood investigations done here showed altered liver enzymes (alanine aminotransferase - 44 U/L, aspartate aminotransferase [AST] - 83 U/L, alkaline phosphatase [ALP] - 595 U/L, A/G - 2.2/3.4), creatinine of 0.8 mg/dl, prothrombin time-international normalized ratio (PT-INR) was elevated (1.86), Hb - 16.9 g/dl, platelet count - 140,000, and urine showed proteinuria (3+).
Esophagogastroduodenoscopy showed Grade-2 esophageal varices with gastropathy and duodenopathy. Computerized tomography (CT) abdomen with contrast [Figure 1], showed hepatomegaly with heterogeneous hepatic parenchymal enhancement with areas of hypodense areas in portal venous phase, homogenizing on delayed images (3 min); nonopacification of the right, middle, and left hepatic veins. Splenomegaly with moderate ascites. The diagnosis of Budd–Chiari syndrome was considered.
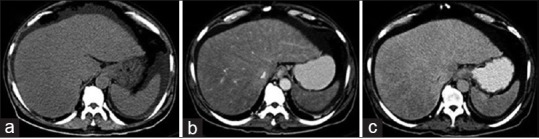
| Figure 1:(a) Plain computerized tomography scan showing hepatosplenomegaly with ascites, (b) arterial phase showing heterogeneous hepatic parenchymal enhancement, (c) venous phase showing areas of hypodense nonenhancing areas in the right lobe of liver with nonopacification of hepatic veins (right, middle, and left) draining into inferior vena cava
However, subsequent liver ultrasound with color Doppler [Figure 2] showed low-velocity flow in hepatic veins and portal veins with no evidence of filling defects to suggest venous thrombosis. Hepatic vein lumen was patent. Due to an elevated PT-INR (1.9) and borderline platelet count (1.4 lakhs), percutaneous liver biopsy was deferred, and transjugular liver biopsy with hepatovenous pressure gradient assessment was considered to rule out any membranous obstruction due to Budd–Chiari syndrome.

| Figure 2:Ultrasound liver with Doppler showing, (a) B-flow imaging showing patent inferior vena cava and left hepatic veins, (b) patent middle hepatic flow, (c) color Doppler images of the right hepatic veins showing hepatofugal patent flow with attenuated phasicity, (d) ultrasonography of liver showing heterogenous echotexture of parenchyma with ascites and cholestasis
Transjugular infrahepatic inferior venacavogram and hepatic venogram showed no evidence of any filling defects/stenosis in inferior vena cava/hepatic veins (right, middle, and left). Hepatovenous pressure gradient assessment showed features suggestive of portal hypertension, and subsequently, a transjugular liver biopsy was done from the right hepatic vein approach, and the sample was sent for histopathological evaluation.
Microscopy showed [Figure 3], liver tissue with distorted spatial relationship between portal tract and central vein. The parenchyma showed linear sinusoidal deposition of amorphous eosinophilic material causing atrophy of liver cell plates. The portal tracts also showed similar deposits. No significant lobular or portal inflammation. The material was congophilic on Congo red stain and showed apple green birefringence on polarizing microscopy. This congophilia persisted after pretreatment with KMnO4. The material was weakly positive for periodic acid-Schiff. With this, a diagnosis of amyloidosis liver, possibly AL type (primary) was made.
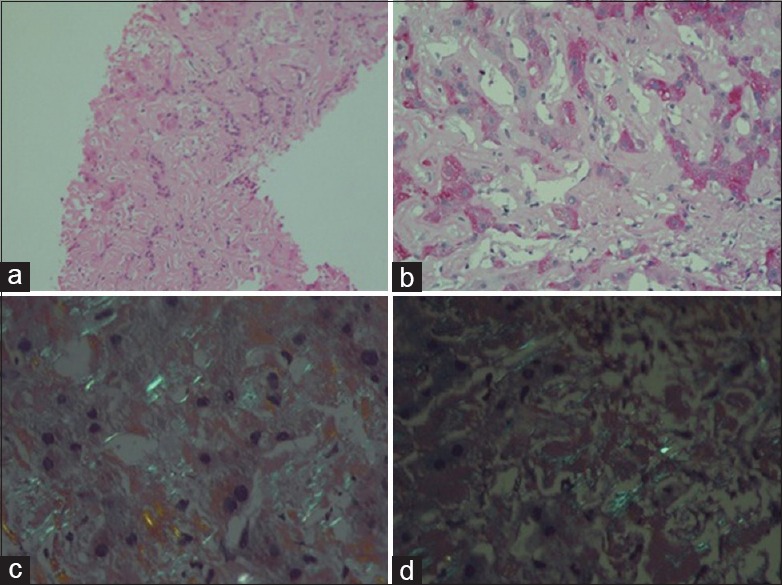
| Figure 3:(a) Liver tissue showing amorphous eosnophilic material in the sinusoids (H and E, ×100), (b) faint periodic acid-Schiff-positive material in the sinusoids (×200), (c) apple green birefringence on polarizing microscopy of Congo red stain (×400), (d) congophilia persists after treatment with KMnO4 (×400)
Serum electrophoresis showed no features to suggest multiple myeloma. The patient was subsequently managed conservatively and planned to be followed up for symptomatic management.
Discussion
Amyloidosis is a multisystemic diffusely infiltrating disease due to deposition of fibrils of protein- mucopolysaccharide complexes. The type of protein deposited determines the subgroup of amyloid.
In primary amyloidosis, light chain immunoglobulin (AL) is deposited.[1] Thus, primary amyloidosis is associated with multiple myeloma, Waldenstrom macroglobulinemia, lymphoplasmacytic lymphoma, etc.[2,3,4] Bone marrow biopsy is required for all patients with AL amyloidosis to exclude overt multiple myeloma. In primary AL amyloidosis, the presence of light chain immunoglobulin in serum with no associated heavy chain is commoner than those associated with an IgM heavy chain which was found in around 4.4%.[5]
In secondary amyloidosis (AA type), protein deposition happens secondary to inflammation; thus, secondary amyloidosis occurs in chronic inflammatory diseases such as rheumatoid arthritis, tuberculosis, Crohn's disease, bronchiectasis, and chronic osteomyelitis.
Third subtype, hereditary or senile type of amyloid (ATTR) occurs due to mutant and wild type of transthyretin.[1]
In localized or organ-limited type, amyloid material is confined to an organ, commonly the respiratory tract, skin, urinary tract, liver, etc. Localized amyloidosis is usually primary (AL type) and rarely presents as secondary (AA type).[6] Localized amyloidosis is diagnosed primarily by excluding other systemic involvement.
In systemic type of disease or secondary amyloidosis, abnormal proteins are deposited in multiple organs, most common is the gastrointestinal tract with colon more frequently involved;[4] other tissues such as myocardium, striated muscle, and adipose tissue, are also involved.
Amyloidosis is usually observed as a systemic form although 10%–20% is localized.[7] Systemic amyloidosis is progressive and fatal, death usually occurs as a result of renal or heart failure.[8] In localized amyloidosis, the management is usually supportive and localized and does not require systemic therapy. Due to therapeutic and prognostic implications, distinction between the primary and secondary type of amyloidosis is clinically vital.[8]
Hepatic amyloidosis
In hepatic amyloidosis, amyloid is deposited in the hepatic parenchyma, within the space of Disse along the sinusoids and/or along the blood vessel walls. Hepatocytes are severely compressed by the accumulation of amyloid material resulting in atrophy or near disappearance of hepatocytes. Advanced cases of amyloid infiltration, results in hepatomegaly with rubbery elastic consistency and may show “lardaceous liver” appearance on the cut surface.[9]
Hepatic involvement in amyloidosis is common, although with mild clinical manifestation.[10] Hepatomegaly with borderline abnormal liver function tests is more frequent findings seen with hepatic amyloidosis.[10] Symptomatic presentations are due to hepatic failure, portal hypertension, and rarely due to organ rupture.[9,11,12]
There has been report that 77% of hepatic amyloidosis is associated with nephrotic syndrome, congestive heart failure, peripheral neuropathy, or orthostatic hypotension.[13] Other signs of hepatic amyloidosis include proteinuria (88%), elevated serum ALP (86%), abnormal serum protein electrophoresis (64%), hyposplenism on peripheral blood smear defined by the presence of Howell–Jolly bodies (62%), hepatomegaly disproportionate to abnormal liver enzymes (81%).[13]
Liver function tests are not sensitive or specific, with a normal level of bilirubin and AST seen in 32% of cases.[13] The median survival of patients with hepatic amyloidosis was 9 months.[14,15]
Diagnosis of hepatic amyloidosis
Radiological findings of hepatic amyloidosis are nonspecific. Ultrasound may show heterogeneous or coarse parenchymal echotexture. Unenhanced CT shows enlarged liver with heterogeneously decreased parenchymal attenuation.[9,16] Parenchymal calcifications are rarely described. On contrast enhanced CT, areas of amyloid infiltration appear as focal hypoattenuating areas, with subsequent contrast filling of these areas in delayed images. The findings are probably due to impaired blood flow as a result of amyloid infiltration of blood vessel walls and hepatocyte congestion.
Magnetic resonance imaging with oral manganese-containing contrast agent revealed several focal areas without contrast uptake by the hepatocytes and no biliary secretion in delayed images at 8 h, with no evidence of any extrahepatic biliary obstruction.[17]
Definitive diagnosis requires histopathological evaluation as diagnostic imaging findings are non specific. Histologically, amyloid is recognized as a homogeneous extracellular material displaying apple green birefringence after positive Congo red staining. It shows aggregation of approximately 10 nm wide fibrils on electron microscopy, exhibits a beta-pleated sheet configuration on radiographic analysis and show resistance to proteases other than pronase.[18]
Conclusion
- Hepatic amyloidosis, clinical presentation, and imaging manifestations are usually varied and nonspecific, which may cause a delay in diagnosis. Biopsy is always required for confirmative diagnosis
- Cases with absent filling of hepatic veins in venous phase should always undergo a hepatic Doppler/B-mode imaging to visualize hepatic vein lumen and demonstrate venous flow. In such cases with contrast enhanced CT showing absent venous filling of hepatic veins, with hepatic Doppler/B-mode imaging showing patent hepatic vein, the possibility of underlying infiltrating pathology of hepatic parenchyma should always be considered.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997;337:898-909.
- Rajkumar SV, Dispenzieri A, Kyle RA. Monoclonal gammopathy of undetermined significance, Waldenström macroglobulinemia, AL amyloidosis, and related plasma cell disorders: Diagnosis and treatment. Mayo Clin Proc 2006;81:693-703.
- Urban BA, Fishman EK, Goldman SM, Scott WW Jr., Jones B, Humphrey RL, et al. CT evaluation of amyloidosis: Spectrum of disease. Radiographics 1993;13:1295-308.
- Georgiades CS, Neyman EG, Barish MA, Fishman EK. Amyloidosis: Review and CT manifestations. Radiographics 2004;24:405-16.
- Gertz MA, Kyle RA, Noel P. Primary systemic amyloidosis: A rare complication of immunoglobulin M monoclonal gammopathies and Waldenström's macroglobulinemia. J Clin Oncol 1993;11:914-20.
- Tirzaman O, Wahner-Roedler DL, Malek RS, Sebo TJ, Li CY, Kyle RA. Primary localized amyloidosis of the urinary bladder: A case series of 31 patients. Mayo Clin Proc 2000;75:1264-8.
- Scott PP, Scott WW Jr., Siegelman SS. Amyloidosis: An overview. Semin Roentgenol 1986;21:103-12.
- Kawashima A, Alleman WG, Takahashi N, Kim B, King BF Jr., LeRoy AJ. Imaging evaluation of amyloidosis of the urinary tract and retroperitoneum. Radiographics 2011;31:1569-82.
- Monzawa S, Tsukamoto T, Omata K, Hosoda K, Araki T, Sugimura K. A case with primary amyloidosis of the liver and spleen: Radiologic findings. Eur J Radiol 2002;41:237-41.
- Kim SH, Han JK, Lee KH, Won HJ, Kim KW, Kim JS, et al. Abdominal amyloidosis: Spectrum of radiological findings. Clin Radiol 2003;58:610-20.
- Gertz MA, Kyle RA. Hepatic amyloidosis: Clinical appraisal in 77 patients. Hepatology 1997;25:118-21.
- Gastineau DA, Gertz MA, Rosen CB, Kyle RA. Computed tomography for diagnosis of hepatic rupture in primary systemic amyloidosis. Am J Hematol 1991;37:194-6.
- Shin YM. Hepatic amyloidosis. Korean J Hepatol 2011;17:80-3.
- Gertz MA, Kyle RA. Hepatic amyloidosis (primary [AL], immunoglobulin light chain): The natural history in 80 patients. Am J Med 1988;85:73-80.
- Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: Clinical features and natural history in 98 patients. Medicine (Baltimore) 2003;82:291-8.
- Mainenti PP, D'Agostino L, Soscia E, Romano M, Salvatore M. Hepatic and splenic amyloidosis: Dual-phase spiral CT findings. Abdom Imaging 2003;28:688-90.
- Møller JM, Santoni-Rugiu E, Chabanova E, Løgager V, Hansen AB, Thomsen HS. Magnetic resonance imaging with liver-specific contrast agent in primary amyloidosis and intrahepatic cholestasis. Acta Radiol 2007;48:145-9.
- Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses ( first of two parts). N Engl J Med 1980;302:1283-92.
| Figure 1:(a) Plain computerized tomography scan showing hepatosplenomegaly with ascites, (b) arterial phase showing heterogeneous hepatic parenchymal enhancement, (c) venous phase showing areas of hypodense nonenhancing areas in the right lobe of liver with nonopacification of hepatic veins (right, middle, and left) draining into inferior vena cava
| Figure 2:Ultrasound liver with Doppler showing, (a) B-flow imaging showing patent inferior vena cava and left hepatic veins, (b) patent middle hepatic flow, (c) color Doppler images of the right hepatic veins showing hepatofugal patent flow with attenuated phasicity, (d) ultrasonography of liver showing heterogenous echotexture of parenchyma with ascites and cholestasis
| Figure 3:(a) Liver tissue showing amorphous eosnophilic material in the sinusoids (H and E, ×100), (b) faint periodic acid-Schiff-positive material in the sinusoids (×200), (c) apple green birefringence on polarizing microscopy of Congo red stain (×400), (d) congophilia persists after treatment with KMnO4 (×400)
References
- Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997;337:898-909.
- Rajkumar SV, Dispenzieri A, Kyle RA. Monoclonal gammopathy of undetermined significance, Waldenström macroglobulinemia, AL amyloidosis, and related plasma cell disorders: Diagnosis and treatment. Mayo Clin Proc 2006;81:693-703.
- Urban BA, Fishman EK, Goldman SM, Scott WW Jr., Jones B, Humphrey RL, et al. CT evaluation of amyloidosis: Spectrum of disease. Radiographics 1993;13:1295-308.
- Georgiades CS, Neyman EG, Barish MA, Fishman EK. Amyloidosis: Review and CT manifestations. Radiographics 2004;24:405-16.
- Gertz MA, Kyle RA, Noel P. Primary systemic amyloidosis: A rare complication of immunoglobulin M monoclonal gammopathies and Waldenström's macroglobulinemia. J Clin Oncol 1993;11:914-20.
- Tirzaman O, Wahner-Roedler DL, Malek RS, Sebo TJ, Li CY, Kyle RA. Primary localized amyloidosis of the urinary bladder: A case series of 31 patients. Mayo Clin Proc 2000;75:1264-8.
- Scott PP, Scott WW Jr., Siegelman SS. Amyloidosis: An overview. Semin Roentgenol 1986;21:103-12.
- Kawashima A, Alleman WG, Takahashi N, Kim B, King BF Jr., LeRoy AJ. Imaging evaluation of amyloidosis of the urinary tract and retroperitoneum. Radiographics 2011;31:1569-82.
- Monzawa S, Tsukamoto T, Omata K, Hosoda K, Araki T, Sugimura K. A case with primary amyloidosis of the liver and spleen: Radiologic findings. Eur J Radiol 2002;41:237-41.
- Kim SH, Han JK, Lee KH, Won HJ, Kim KW, Kim JS, et al. Abdominal amyloidosis: Spectrum of radiological findings. Clin Radiol 2003;58:610-20.
- Gertz MA, Kyle RA. Hepatic amyloidosis: Clinical appraisal in 77 patients. Hepatology 1997;25:118-21.
- Gastineau DA, Gertz MA, Rosen CB, Kyle RA. Computed tomography for diagnosis of hepatic rupture in primary systemic amyloidosis. Am J Hematol 1991;37:194-6.
- Shin YM. Hepatic amyloidosis. Korean J Hepatol 2011;17:80-3.
- Gertz MA, Kyle RA. Hepatic amyloidosis (primary [AL], immunoglobulin light chain): The natural history in 80 patients. Am J Med 1988;85:73-80.
- Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: Clinical features and natural history in 98 patients. Medicine (Baltimore) 2003;82:291-8.
- Mainenti PP, D'Agostino L, Soscia E, Romano M, Salvatore M. Hepatic and splenic amyloidosis: Dual-phase spiral CT findings. Abdom Imaging 2003;28:688-90.
- Møller JM, Santoni-Rugiu E, Chabanova E, Løgager V, Hansen AB, Thomsen HS. Magnetic resonance imaging with liver-specific contrast agent in primary amyloidosis and intrahepatic cholestasis. Acta Radiol 2007;48:145-9.
- Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses ( first of two parts). N Engl J Med 1980;302:1283-92.