 PDF
PDF  Views
Views  Share
Share


Follicular dendritic cell sarcoma with paraneoplatic pemphigus: Rare case and a brief review of literature
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2013; 34(04): 317-319
DOI: DOI: 10.4103/0971-5851.125255
Abstract
Paraneoplastic pemphigus (PNP) is often a fatal autoimmune bullous disease characterized by severe stomatitis, polymorphous skin eruptions, and underlying neoplasms. We describe a patient with PNP associated with follicular dendritic cell sarcoma (FDCS), a rare neoplasm originating from follicular dendritic cells, which are non-lymphoid, non-phagocytic accessory cells of the lymphoid system and play an integral role in regulation of the germinal center reaction and present antigens to B-cells. The presence of rich vascularity around the tumor and few hyalanized vascular follicles found in histopathological examination gives the clue that the tumor might have developed from Castleman′s disease (CD). As for the mechanisms by which CD induces PNP, it has been proposed that autoantibodies secreted from the Castleman′s tumor play pivotal role. This hypothesis seems to be supported by the present case, in which CD may have triggered both the FDCS and the PNP.
Publication History
Article published online:
19 July 2021
© 2013. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Paraneoplastic pemphigus (PNP) is often a fatal autoimmune bullous disease characterized by severe stomatitis, polymorphous skin eruptions, and underlying neoplasms. We describe a patient with PNP associated with follicular dendritic cell sarcoma (FDCS), a rare neoplasm originating from follicular dendritic cells, which are non-lymphoid, non-phagocytic accessory cells of the lymphoid system and play an integral role in regulation of the germinal center reaction and present antigens to B-cells. The presence of rich vascularity around the tumor and few hyalanized vascular follicles found in histopathological examination gives the clue that the tumor might have developed from Castleman's disease (CD). As for the mechanisms by which CD induces PNP, it has been proposed that autoantibodies secreted from the Castleman's tumor play pivotal role. This hypothesis seems to be supported by the present case, in which CD may have triggered both the FDCS and the PNP.
INTRODUCTION
Follicular dendritic cell sarcoma (FDCS) is a rare neoplasm (0.4% of soft tissue sarcomas)[1] originating from follicular dendritic cells (FDCs), a nonlymphoid, nonphagocytic antigen presenting cells of the lymphoid system.[2] The tumor was first characterized by Monda[3] et al., and subsequently, specific immunohistochemical markers[2] have been identified. Since that time, only 3 cases of FDCS arising from castleman's disease (CD) with paraneoplastic pemphigus (PNP) have been reported in the literature.[4,5,6]
CASE REPORT
A 20-year-old boy presented with 7 months history of multiple polymorphous vesiculobullous mucocutaneous eruptions around the lips and subsequently in buccal mucosa causing dysphagia. He later developed conjuntivitis associated with blurred vision and atypical targetoid lesions with crusts surrounded by erythema scattered on the trunk and extremities including prepuce and perianal region [Figure 1]. He lost 5 kg during this period. Direct immunoflorescence of biopsy specimen from Skin lesion showed deposition of Immunoglobulin G (+) and C3 in the intercellular region of epidermal cells mainly in the suprabasal region. Linear deposition of IgG is also seen in dermo-epidermal junction suggestive of PNP. Computed tomography (CT) scan revealed a well defined intensely enhancing mass lesion with central necrosis in the right hemipelvis, 6 cm in size, abutting the right obturator internus muscle and displacing the urinary bladder anteriorly. Engorged vessels were seen around the mass [Figure 2]. Exploratory laprotomy was done but the mass was found to be unresectable. Histopathlogy of the mass showed oval to spindle cells in a background of vascular stroma infiltrated by lymphocytes. The cells were immunopsitive for CD21, CD35, CD23, S-100, Claustrin, CD34, CD31, S-100; Leucocyte common antigen, and H uman leukocyte antigen-DR were also positive in some cells. Hyaline-vascular follicles were identified at the peripheral portion or occasionally scattered in the center of the tumor. Rich vascularity was highlighted by the CD34 immunostaining [Figures [Figures33 and and4].4]. Overall features favoured FDCS. The presence of rich vascularity around the tumor and few hyalanized vascular follicles found in histopathological examination (HPE) gave the clue that the tumor might have developed from CD. He tested negative for human immunodeficiency virus. He was started on systemic steroids. His truncal lesions marginally improved after steroids but there was no improvement in arm lesions. He was then given Rituximab, cyclophosphamide, Adriamycin, vincristin, prednisolone. He expired post 1st course of chemotherapy due to pseudomonas infection.
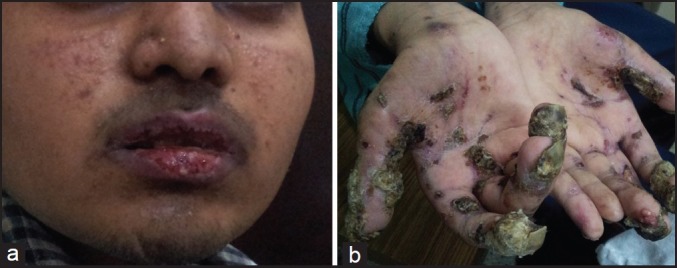
| Fig. 1 (a) Vesiculobullous mucocutaneous eruptions around the lips (b) Crusts surrounded by erythema scattered on the extremities
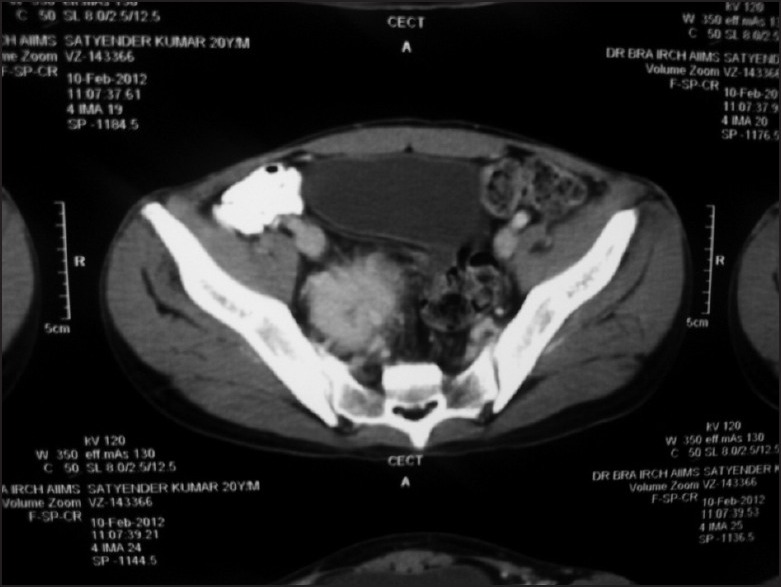
| Fig. 2 Well defined intensely enhancing mass lesion with central necrosis in the right hemipelvis, 6 cm in size, abutting the right obturator internus muscle and displacing the urinary bladder anteriorly
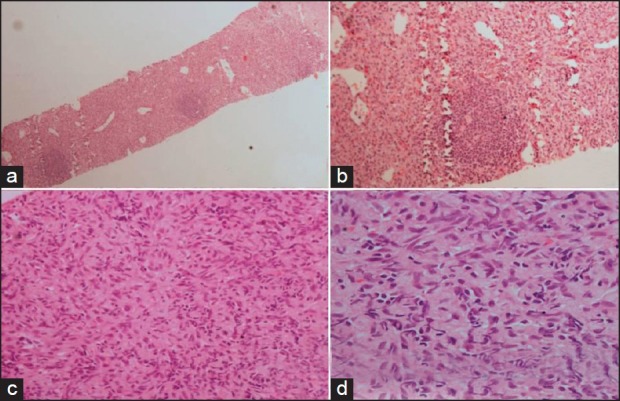
| Fig. 3 Photomicrograph shows cells are arranged in sheets with small lymphocytic nodules forming germinal center. [Fig a, H&E ×40 and b ×100]. The tumor cells are oval to slindle with ill defined border. The nuclei are oval to spindle with vesicular chromatin. [Fig c, ×200, d ×400]
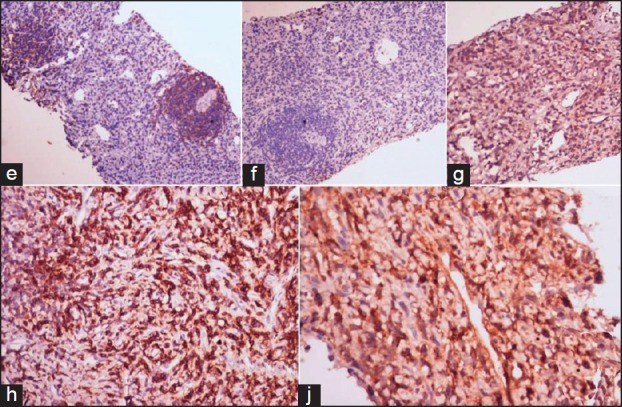
| Fig. 4 Photomicrograph shows positivity of CD20 in the nodules [Fig 4e, IHC-CD20 ×100], the nodules are negative for CD3 (4f, ×100), the spindle cells are immunopositive for CD35, CD43, Clusterin [Fig 4g, IHC-CD35 ×100, h-CD43 ×200, j-Clusterin ×200]
DISCUSSION
FDCS arises from FDCs which are localized within lymphoid follicles; morphologically have entangled cytoplasmic processes and frequent desmosomes; have the ability to trap and retain antigens for long periods of time in the form of immune complexes, but have a non-phagocytic activity; and express molecules involved in the proliferation and differentiation of B-cells.[2] The only predisposing factor identified for FDCS is hyaline-vascular CD (10-20% of cases).[7] FDCS are associated with Epstein barr virus (12%) especially those occurring in the liver.[7] It generally affect young to middle-aged adults, with a mean age of 43 years with no sex predilection.[8] They usually present as painless coalescent nodal mass (60%) of variable size[8] (1-20 cm diameter), but extranodal disease is most commonly seen in the head and neck (tonsil, nasopharynx) and intraabdominal organs (in particular the pancreas and peripancreatic tissues).[7] Other locations: axilla, gastrointestinal tract (17%), liver, oral cavity (13%), mediastinum (8%) and spleen (2%).[7]
Grossly it appears as a firm white mass. HPE showed ovoid to spindled cells whorls with long cellular processes, distinctive nuclei, speckled to vesicular chromatin; sometimes multinucleated cells. Hemorrhage or necrosis is an uncommon finding.[7] Immunohistochemistry positive for CD21, CD23, CD35, Fascin, Clusterin, podoplanin, Desmoplakin Epithelial membrane antigen, Epithelial growth factor receptor, Anti-Follicular Dendritic Cells antibody(Ki-M4), Anti-Follicular Dendritic Cells antibody CNA 42, CD68, Fragment crystallizable region, S100 ± (but not strong) Factor XIIIa ± while negative for Leucocyte common markers (CD1a), cytokeratin, lysozyme, CD34, CD3, CD79a, CD30, Human Melanoma Black HMB45.[2]
A subset of these tumors has been reported to arise in or concomitantly with foci of CD[7] as in our case. The presence of rich vascularity around the tumor and few hyalanized vascular follicles found in HPE gave the clue that the tumor might have developed from CD. In fact, some argue that FDCS actually develops from CD as a hyperplasia — dysplasia — neoplasia sequence[7] so FDCS may share some of the radiologic features of CD. In our patient, we saw a large lobulated soft-tissue mass with engorged vessels seen around it on CT, the feature of CD.
FDCS behaves more like low-grade sarcoma than lymphoma. Complete surgical resection is considered the treatment of choice; the role of adjuvant therapy (chemotherapy or radiation) is unclear because of the rarity of the disease. Cyclophosphamide, Adriamycin, vincristin, prednisolone CHOP[9] have shown good response. In a small series by Soriano et al.[9] patients who received all three modalities of treatment upfront had the longest disease-free intervals. Sarcoma-based regimen gemcitabine and taxane;[9] imatinib in combination with gemcitabine and cisplatin,[10] rituximab[11] was also reported to have some activity against FDCS. Our patient was given R-CHOP. But response could not be assessed as he expired post first cycle due to pseudomonas infection.
Poor prognostic factors like extensive necrosis, a mass larger than 6 cm, cytologic atypia, intraabdominal location, and a high proliferative index increase the likelihood of recurrence, metastasis, and mortality.[7] The lung, liver, and lymph nodes are the most common sites for metastasis, and the tumor may be highly aggressive and fatal within 2 years of diagnosis.[7] Local recurrence rate was reported to be 43% while distant metastases – 24% (lung, liver, peritoneum, pancreas and lymph nodes). Five-year recurrence-free survival rates of 27% to 32%. And overall 5-year survival rate of 79%.[7]
Our patient presented with PNP, an autoimmune bullous disease charaecterized by severe stomatitis, polymorphous skin eruptions that occurs in association with various forms of underlying neoplasia. It is characterized by the presence of autoantibodies that react with intermediate filament-associated proteins in desmosomes and hemidesmosomes; desmoplakin, bullous pemphigoid antigen 1, and envoplakin.[4]
A review of 163 case reports of PNP reported that 84% of cases were associated with hematologic malignancies (Non hodgkin lymphoma [39%], Chronic lymphocytic leukemia [18%], CD [18%], thymoma [6%], Waldenstrom macroglobulinemia [1%], Hodgkin's lymphoma [1%], and monoclonal gammopathy [1%]). The remaining 16% were associated with nonhematologic neoplasms (epithelial origin carcinoma [9%], mesenchymal origin sarcoma [6%], and melanoma [1%]). Oral ulcerations were the presenting feature in 45% of all cases and appear to be a key feature of PNP.[12]
In conclusion, our case report is unique because it reports for the 1st time to our knowledge as a pathological constellation of follicular dendritic cell sarcoma with castleman's disease and paraneoplastic pemphigus arising from extranodal pelvic region, a rare disease at a rare site. Its importance lies in suspicion of diagnosis and considering the treatment of paraneoplastic pemphigus according to the underlying pathology. Being rare its optimal treatment yet to be defined.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 (a) Vesiculobullous mucocutaneous eruptions around the lips (b) Crusts surrounded by erythema scattered on the extremities
| Fig. 2 Well defined intensely enhancing mass lesion with central necrosis in the right hemipelvis, 6 cm in size, abutting the right obturator internus muscle and displacing the urinary bladder anteriorly
| Fig. 3 Photomicrograph shows cells are arranged in sheets with small lymphocytic nodules forming germinal center. [Fig a, H&E ×40 and b ×100]. The tumor cells are oval to slindle with ill defined border. The nuclei are oval to spindle with vesicular chromatin. [Fig c, ×200, d ×400]
| Fig. 4 Photomicrograph shows positivity of CD20 in the nodules [Fig 4e, IHC-CD20 ×100], the nodules are negative for CD3 (4f, ×100), the spindle cells are immunopositive for CD35, CD43, Clusterin [Fig 4g, IHC-CD35 ×100, h-CD43 ×200, j-Clusterin ×200]