 PDF
PDF  Views
Views  Share
Share


Fanconi anemia presenting as an “evolving” acute leukemia-diagnostic challenges
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2013; 34(04): 305-308
DOI: DOI: 10.4103/0971-5851.125251
Abstract
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous recessive disorder characterized by diverse congenital malformations, progressive pancytopenia and predisposition to both hematologic malignancies and solid tumors. We report, a 14-year-old boy who presented with clinical features of aplastic anemia (AA). Subsequent bone marrow examination and multiparametric flowcytometric immunophenotyping revealed an evolving hypoplastic acute myeloid leukemia. Chromosomal breakage studies using clastogenic agent mitomycin C showed 88% stress induced chromosomal/chromatid breaks, gaps and rearrangements revealing an underlying FA. The case emphasizes upon the role of a systematic clinico-investigative approach in diagnosing such patients who by clinical criteria appear to have idiopathic AA and appear phenotypically normal. A timely and accurate diagnosis becomes vital in these cases to implement appropriate therapy.
Keywords
Acute myeloid leukemia - chromosomal breaks fanconi - cytogenetics - flowcytometric immunophenotyping - hypocellular - mitomycin CPublication History
Article published online:
19 July 2021
© 2013. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous recessive disorder characterized by diverse congenital malformations, progressive pancytopenia and predisposition to both hematologic malignancies and solid tumors. We report, a 14-year-old boy who presented with clinical features of aplastic anemia (AA). Subsequent bone marrow examination and multiparametric flowcytometric immunophenotyping revealed an evolving hypoplastic acute myeloid leukemia. Chromosomal breakage studies using clastogenic agent mitomycin C showed 88% stress induced chromosomal/chromatid breaks, gaps and rearrangements revealing an underlying FA. The case emphasizes upon the role of a systematic clinico-investigative approach in diagnosing such patients who by clinical criteria appear to have idiopathic AA and appear phenotypically normal. A timely and accurate diagnosis becomes vital in these cases to implement appropriate therapy.
INTRODUCTION
Fanconi anemia (FA) is an autosomal recessive (AR) chromosomal instability disorder, belonging to more than one class of cancer predisposition syndromes i.e. AR disorders of deoxyribonucleic acid (DNA) repair as well as inherited bone marrow failure syndromes. It is characterized by congenital abnormalities, defective hemopoiesis and high risk of developing acute myeloid leukemia (AML) and certain solid tumors. FA is found in all races and ethnic groups. Its overall prevalence is 1 to 5 cases/million with a carrier frequency of 1:200 to 300 in most populations.[1,2] These estimates are based on the incidence of affected individuals before the full spectrum of the FA phenotype was recognized. The true gene frequency is likely to be markedly higher than this. We report, a case of FA presenting with an “evolving” AML to emphasize upon the importance of a timely and comprehensive clinico-investigative work-up.
CASE REPORT
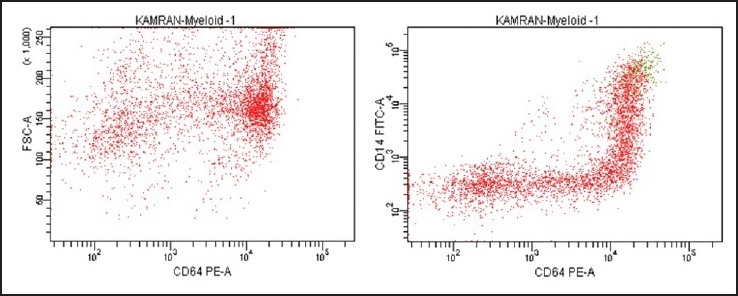
A 14-year-old male patient presented with the complaints of generalized weakness and off and on high grade fever for 6 months. On examination, he was well-looking, afebrile and had pallor without any evidence of mucocutaneous bleeding. There was no lymphadenopathy or organomegaly. His complete blood counts (CBC) showed a hemoglobin (Hb) of 8.7 g/dl, total leucocyte count (TLC) of 1.5 × 109/L and a platelet count of 38 × 109/L. Red cell indices showed hematocrit of 25.2 %, mean corpuscular volume of 102.5 fl, mean corpuscular Hb of 35.3 pg and mean corpuscular Hb concentration of 34.4 g/dl. A clinical diagnosis of aplastic anemia (AA) was made and bone marrow examination was carried out. The marrow aspirate smears were hemodilute and pauci-particulate with very few marrow cell trails showing presence of blast cells, mature monocytes and cells with promonocytoid features (~10%, ~13% and ~4% of the evaluable marrow cells, respectively) [Figure 1]. Erythroid cells showed megaloblastic erythropoiesis with dyserythropoiesis. There was a prominence of macrophages with hemophagocytosis. The blast cells were negative for myeloperoxidase (MPO) cytochemical staining. Bone marrow trephine biopsy was suboptimal and insufficient for opinion. In view of the hemodilution and lack of appreciable dyshemopoietic features, a diagnosis of an “evolving” MPO negative acute leukemia was given and flowcytometric immunophenotyping (FCIM) was suggested for further evaluation. Meanwhile, the clinical course of patient was apparently uneventful except for an acute onset submandibular swelling that subsided with antibiotic therapy. His subsequent CBC at next presentation after 2 weeks showed persistent pancytopenia with Hb of 10.2 g/dl, TLC of 2.3 × 109/L and 21 × 109/L platelets. At this time, his marrow aspirate smears showed hypocellular marrow cell trails with myelogram comprising 18% blasts, 10% promonocytes, 04% monocytes, 10% polymorphs, 01% metamyelocytes, 03% eosinophils, 42% lymphocytes, 04% plasma cells and 08% erythroid precursor cells [Figure 2]. A diagnosis of hypocellular marrow with AML was made. The conventional cytogenetics revealed a normal karyotype [Figure 3]. At subsequent presentation, the patient was apparently maintained except for worsening anemia and thrombocytopenia. There was no bleeding and his clinical examination did not reveal any new findings. His peripheral blood was sent for the stress cytogenetics and human leukocyte antigen (HLA) typing. However, after 3 months from initial presentation, he came with high grade fever and bone pains and finally consented for admission. His CBC showed 6.6 g/dl Hb, 0.8 × 109/L leucocytes and 3 × 109/L platelets. Bone marrow aspirate revealed hypocellular particles with ~68% blasts and promonocytes. ~3% blast cells showed MPO positivity. The 6-color FCIM (FACS Canto II, BD Biosciences) on bone marrow revealed significant replacement by a prominent cell population (red) with variable CD45 expression. This abnormal cluster spanned from the blast region into the monocyte position with bulk of the neoplastic cells falling into the medium cell size range [Figure 4a]. A small number of lymphocytes (blue), few residual myeloid precursors (purple) as well as maturing monocytic elements (green) were also present. The leukemic cells comprised ~45% of the cells in the sample and showed homogeneously bright expression of CD33, variable expression of CD13, CD34 and displayed a complex pattern of CD14/CD64 co-expression, indicating monocytic maturation [Figure [Figure4b4b and andc].c]. These blast cells also showed heterogeneous expression of CD11b, CD15, CD4 and bright HLA DR [Figure 4d]. The maturing monocytic elements (comprising ~3% of cells in the sample) showed normal expression of brighter CD33, CD16, CD13, HLA DR and CD11b. Based on the shape of the abnormal cell cluster on Side Scatter/CD45 dot-plot and immunophenotypic findings including pattern of CD14/CD64 co-expression, diagnosis of AML with monocytic differentiation was made. Stress cytogenetics [Table 1] showed 88% chromosome breaks and rearrangements in patient's peripheral lymphocytes with mitomycin C (MMC) [Figure 5]. The diagnosis of FA was thus established. Patient consequently received packed red cell and platelet support and was started on a low dose cytosine arabinoside 100 mg/M2/daily (adverse drug event induction therapy). Subsequently, he developed typhlitis and was conservatively managed until he left the hospital care against medical advice.
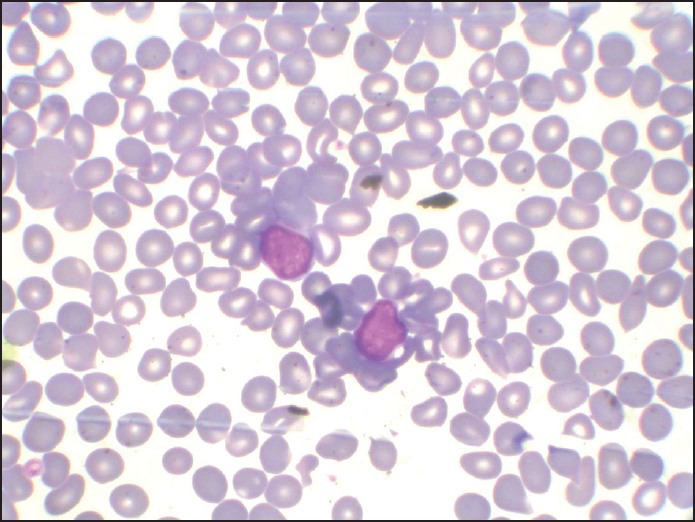
| Fig. 1 H and E-stained marrow aspirate smear (×100) showing prominence of blast cells despite hemodilution
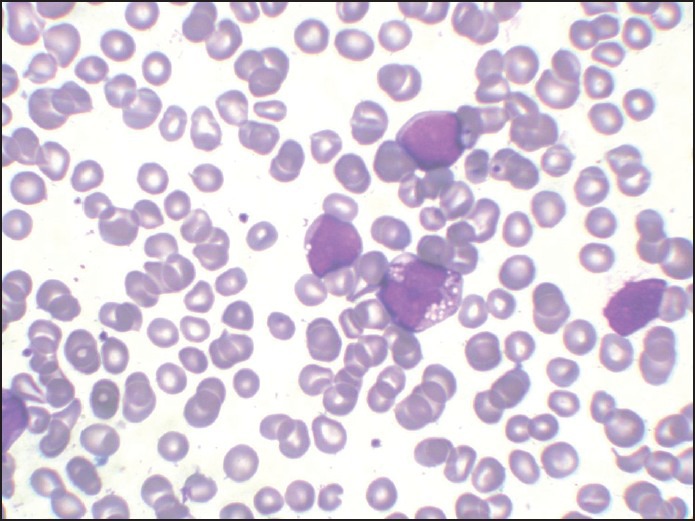
| Fig. 2 H and E-stained marrow aspirate smear (×100) showing acute myeloid leukemia with monoblasts and promonocytes
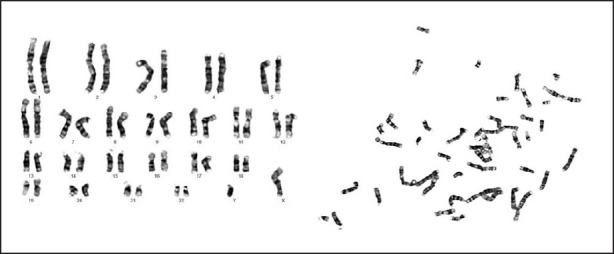
| Fig. 3 Conventional cytogenetics showing a normal karyotype on GTG banding
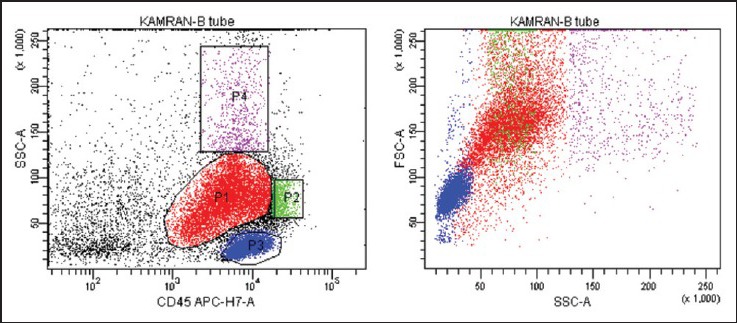
| Fig. 4a 6-color flowcytometric immunophenotyping shows blast cell population (red) extending up to the monocytic region, with variable CD45 expression. The bulk of the neoplastic cells falls into the medium cell size range
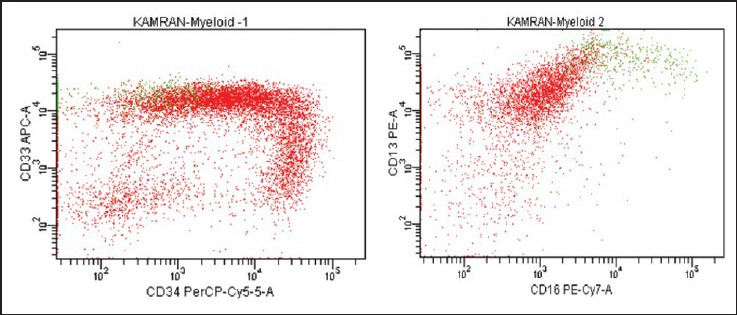
| Fig. 4b CD 13 expression is variable while CD 33 expression is homogeneously bright
| Fig. 4c The “trail” pattern of CD64/CD14 co-expression corresponds to monocytic cells at various stages of maturation. CD14 intensity varies from negative to bright indicating spectrum of immature to mature monocytic cells. Bulk of the blast population shows bright CD64
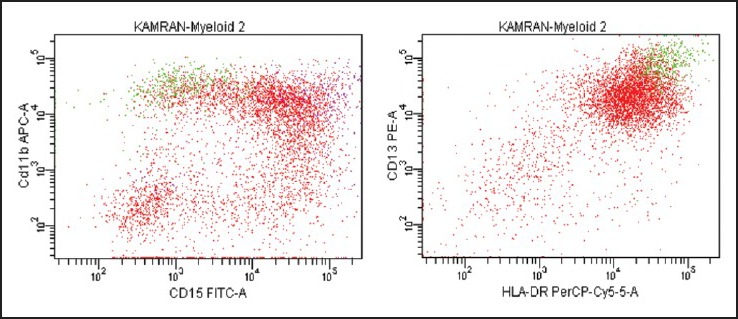
| Fig. 4d CD11b distribution on blast cells is markedly heterogeneous and overlaps with that on residual myeloid precursors (purple). Also note the variability in CD15 expression, imparting a cloud appearance to the blast population on CD11b/CD15 dot-plot. Human leukocyte antigen DR expression is brighter on maturing monocytic elements (green) than blast cells
Table 1
Clastogenic† stress induced chromosomal breakage analysis

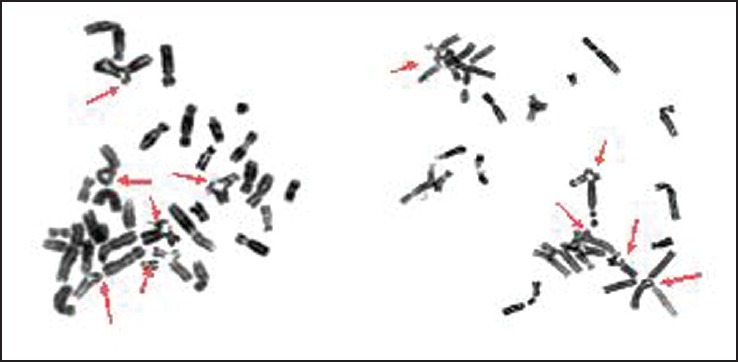
| Fig. 5 Two partial metaphases using clastogenic agent mitomycin C showed chromosomal breaks, gaps and rearrangements
DISCUSSION
Hematologic abnormalities occur in virtually all patients with FA at a median age of 7 years (range: Birth to 41 years).[3,4] Based on clinical data in the International FA registry (IFAR), the initial hematological findings were diverse and some patients presented with myelodysplastic syndrome (MDS) or AML without prior diagnosis of AA. Of the 754 IFAR FA patients, 120 (16%) patients experienced MDS and/ or AML. The cumulative incidence of MDS/ AML by age 40 years was 33%.[4] Based on a survey of FA patients performed by Rosenberg et al., the median age of onset of leukemia was 11.3 years.[5]
Our case had no significant phenotypic abnormalities and his pancytopenia was detected during an episode of fever. The marrow examination caught upon the increase in blast cells despite hemodilution and reduced overall marrow cellularity indicating an evolving acute leukemia in a hypocellular bone marrow. The diagnostic difficulty encountered was largely attributed to the differentiation between pediatric MDS and acute leukemia. The gradual evolution of the disease, lack of significant dyshemopoietic features and fair clinical preservation of patient added to the diagnostic dilemma.
MDS in FA patients is known to behave differently than primary MDS in non-FA patients. Patients with FA may have MDS for long periods of time, with clonal fluctuations and without leukemic transformation. The term MDS is often used if abnormal cytogenetic marrow clones are observed regardless of marrow morphology and is not assumed in the context of FA that MDS represents an obligate precursor to leukemia.[6] In fact, MDS and AML must be distinguished in etiologic researches to determine whether some forms of MDS are preleukemic in FA patients.
On the other hand, FA-related leukemias differ from leukemia in the general population in several important ways. As per surveillance, epidemiology and end results cancer statistics review, 94% of the leukemias were myeloid and only 6% were lymphoid compared with 84% lymphoid leukemias in non-FA children. The age distribution of leukemia in FA patients was normally distributed around a mode of 14 years, in contrast to the general population, in which the incidence of AML is higher in infants, declines around age 10 and then rises slightly in the late teens.[7] Besides, AMLs derived from FA show greater sensitivity to chemotherapy.
One should realize that available studies in the literature are not mutually exclusive; each used different data analysis methods and is prone to potential biases. However, on the basis of these studies the cumulative crude risks (irrespective of age) for MDS are around 5% and 5-10% for leukemia.[8]
The unique biological behavior of hematologic malignancies in FA is clinically relevant and the fact that consideration of FA in the differential diagnosis of a patient mainly depends on the clinician's concept of the FA phenotype, cannot be overemphasized. According to the literature, patients who developed leukemia or MDS or solid tumors were significantly older at the time of diagnosis of FA than patients without these complications.[6] This finding suggests that patients with those events had a milder phenotype and therefore were not diagnosed until they sought medical attention for an adverse event.
The gradual evolution of acute leukemia was initially picked up on marrow morphology and later on confirmed by flowcytometric studies. Chromosomal breakage studies involving clastogenic effect of DNA crosslinking agents i.e. MMC or diepoxybutane are unique diagnostic markers for FA and are especially used to identify pre-anemic cases as well as patients with AA or leukemia who do not have characteristic physical findings.[9,10]
Our case highlights the importance of a complete diagnostic work-up in such patients, who by clinical criteria appear to have idiopathic AA and appear phenotypically normal.
ACKNOWLEDGMENT
The authors would like to thank Dr. Meena Lall (Senior Consultant, Department of Genetic Medicine, Sir Ganga Ram Hospital) for the technical help in Chromosomal Breakage Studies.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 H and E-stained marrow aspirate smear (×100) showing prominence of blast cells despite hemodilution
| Fig. 2 H and E-stained marrow aspirate smear (×100) showing acute myeloid leukemia with monoblasts and promonocytes
| Fig. 3 Conventional cytogenetics showing a normal karyotype on GTG banding
| Fig. 4a 6-color flowcytometric immunophenotyping shows blast cell population (red) extending up to the monocytic region, with variable CD45 expression. The bulk of the neoplastic cells falls into the medium cell size range
| Fig. 4b CD 13 expression is variable while CD 33 expression is homogeneously bright
| Fig. 4c The “trail” pattern of CD64/CD14 co-expression corresponds to monocytic cells at various stages of maturation. CD14 intensity varies from negative to bright indicating spectrum of immature to mature monocytic cells. Bulk of the blast population shows bright CD64
| Fig. 4d CD11b distribution on blast cells is markedly heterogeneous and overlaps with that on residual myeloid precursors (purple). Also note the variability in CD15 expression, imparting a cloud appearance to the blast population on CD11b/CD15 dot-plot. Human leukocyte antigen DR expression is brighter on maturing monocytic elements (green) than blast cells
| Fig. 5 Two partial metaphases using clastogenic agent mitomycin C showed chromosomal breaks, gaps and rearrangements