 PDF
PDF  Views
Views  Share
Share


Extrarenal Extracranial Rhabdoid Tumor of the Pelvis in a Young Adult management of a Challenging Case
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(03): 383-386
DOI: DOI: 10.4103/ijmpo.ijmpo_108_17
Abstract
Rhabdoid tumor commonly occurs in the kidney and has an aggressive clinical course with high mortality. Central nervous system is the most common extrarenal site. Extrarenal extracranial rhabdoid tumor (EERT) is rare. EERT usually presents in childhood, and presentation in adulthood is extremely rare. This tumor is often difficult to diagnose, and there is no established standard of care due to the paucity of cases. We herein report a case of extrarenal rhabdoid tumor of the pelvis in a young adult and discuss the presentation and possible treatment options of this rare tumor.
Publication History
Article published online:
04 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Rhabdoid tumor commonly occurs in the kidney and has an aggressive clinical course with high mortality. Central nervous system is the most common extrarenal site. Extrarenal extracranial rhabdoid tumor (EERT) is rare. EERT usually presents in childhood, and presentation in adulthood is extremely rare. This tumor is often difficult to diagnose, and there is no established standard of care due to the paucity of cases. We herein report a case of extrarenal rhabdoid tumor of the pelvis in a young adult and discuss the presentation and possible treatment options of this rare tumor.
Rhabdoid tumor of the kidney (RTK) is a rare variant of Wilms tumor accounting for 1.6%–2% of childhood renal tumors and has a dismal prognosis.[1] Outside the kidney, it commonly involves central nervous system as atypical teratoid/rhabdoid tumor. Rarely reported extrarenal sites include liver, heart, abdomen, brachial plexus, neck, perineum, and paraspinal regions with a predilection for mediastinum and paraspinal areas.[2] Extrarenal rhabdoid tumor (ERRT) is a deadly disease, with a 3-year survival rate of only 9%.[1] In general, these tumors occur in childhood with a median age of 11–18 months.[3] Presentation in adulthood is rare. We herein report a case of a 20-year-old male patient with pelvic rhabdoid tumor treated with multimodality approach and review the pertinent medical literature.
A 20-year-old male patient presented with a history of pain in the back, radiating down to his lower limbs for 2 years, urinary hesitancy for 1 year, and gradually increasing swelling in the left groin for 4 months. Neurological examination showed limping gait, wasting of the left lower limb muscles with an associated decrease in the power in left lower limb (left hip-grade 3/5, left knee flexors, and extensors-grade 3/5 and 0/5, respectively, left ankle-grade 2/5). Baseline magnetic resonance imaging of the pelvis, done 6 months before presentation at our institute, showed an expansile lesion of size 7.6 cm × 6.6 cm × 8.6 cm involving S2–S3 segment of sacral vertebrae with the involvement of the left sacroiliac joint and iliac bone with large intra-pelvic soft tissue component. The mass was obliterating adjacent sacral neural foramen and compressing intra-pelvic neurovascular bundle, left piriformis muscle, and sciatic nerve. The patient had been diagnosed at a local center with bone Tuberculosis and tubercular abscess and was started on anti-tubercular therapy (ATT), which led to symptom progression. Core biopsy from the sacral mass, performed at our institute, revealed a malignant tumor composed of pleomorphic cells and rhabdoid cells with abundant eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, intracytoplasmic juxta-nuclear eosinophilic inclusion, frequent mitoses, and large areas of necrosis. On immunohistochemistry, the tumor cells stained positive for cytokeratin, epithelial membrane antigen, vimentin, synaptophysin, MIC-2 (CD-99) and negative for smooth muscle actin, myogenin, glial fibrillary acidic protein, Bcl-2, CD-34, and Brachyury. There was a loss of expression of INI-1 in the nuclei of tumor cells. The morphological and immunohistochemical features of the tumor were consistent with a diagnosis of extrarenal rhabdoid tumor [Figure 1]. Bone marrow biopsy and whole body bone scan did not show any evidence of metastasis. A contrast-enhanced computed tomography (CECT) scan of chest, abdomen, and pelvis showed an increase in the size of the lesion (16 cm × 10 cm × 11 cm) with the tumor infiltrating the left psoas muscle, destroying the left side of sacrum, extending extradurally through the S1–S2 sacral foramen and also into the left gluteal and inguinal regions [Figure [Figure2a2a and andb].b]. There was a suspicious nodule in the lower lobe of the right lung suggestive of metastasis [Figure 2c]. Both kidneys were normal. CECT of the brain was essentially normal.
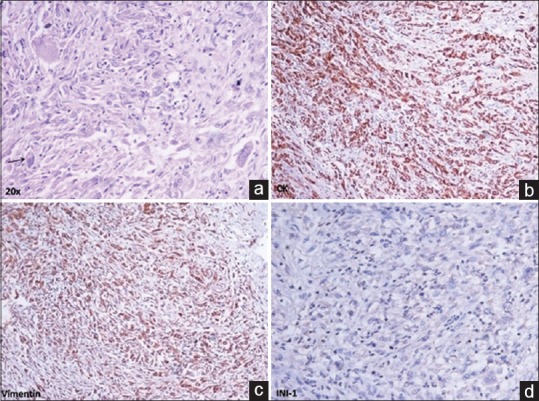
| Figure 1:(a) Malignant tumor composed of pleomorphic cells and rhabdoid cells with vesicular nuclei, prominent nucleoli and intracytoplasmic eosinophilic inclusion (arrow). Immunohistochemistry shows (b) diffuse strong expression of cytokeratin, (c) vimentin and (d) loss of expression of INI-1 in nuclei of tumor cells

| Figure 2:(a and b) Computed tomography scan of pelvis with oral and intravenous contrast showing a lesion arising from the left side of sacrum with involvement of left sacro-iliac joint and left iliac bone, extradural extension through S1–S2 sacral foramina, intrapelvic soft tissue extension, infiltration of the left psoas muscle, and extension into the left groin; (c) computed tomography scan of thorax-lung window showing a suspicious nodule in lower lobe of right lung suggestive of metastasis; (d) contrast-enhanced computed tomography scan of abdomen showing an ill-defined lesion in segment 4a of liver suggestive of metastasis
In view of unresectable tumor and lung metastasis, he received 6 cycles of combination chemotherapy with ICE regimen (injection ifosfamide – 2 g/m2 intravenous [IV] D1–D3 with mesna uroprotection, injection carboplatin – 500 mg/m2 IV D3 and injection etoposide – 100 mg/m2 IV D1–D3), repeated every 3 weeks. Interim CECT scan of chest, abdomen and pelvis, done after 3 cycles of chemotherapy showed a 40%-decrease in the size of mass with resolution of the gluteal component suggestive of partial response. End-of-chemotherapy CECT scan of chest, abdomen, and pelvis showed a further 20%- decrease in the size of the primary lesion with the appearance of a new lesion in segment 4a of liver suggestive of liver metastasis [Figure 2d]. The patient was considered as unsuitable for curative surgery and palliative radiotherapy to the left hemipelvis to a dose of 20 Gray in 5 fractions over 1 week was planned by anterior and posterior fields with 6 MV X-rays on a medical linear accelerator (Varian CL 2300 C/D). The patient received 16 Gray in 4 fractions and then defaulted due to personal reasons. There was 50% subjective improvement 1 month after completion of radiotherapy in the form of improvement of muscle strength in the left lower limb (power-grade 4/5) and decrease in difficulty in walking. Subsequently, he was started on metronomic chemotherapy with a 4 drug combination of tablet celecoxib (200 mg BD), Tab thalidomide (100 mg HS) and alternating courses of tablet cyclophosphamide (50 mg OD) and tablet etoposide (50 mg OD) for 3 weeks each. Three months after the completion of palliative radiotherapy, there was good symptom control and CECT scan of the chest, abdomen and pelvis showed a further decrease (<20>
The rhabdoid tumor was initially described by Beckwith and Palmer as an aggressive, rhabdosarcomatoid variant of the classical renal Wilms tumor.[4] Later, it was identified as a distinct clinicopathologic entity by Haas et al. in 1981.[5] Extrarenal rhabdoid tumors are rare tumors that occur outside the kidney. About 15 cases of ERRT are diagnosed each year in North America.[1] In a study from Childrens Hospital Los Angeles conducted between 1983 and 2003, only 14 cases of extracranial malignant rhabdoid tumor (MRT) were found, of which only one was in the pelvis.[6] Most cases of extrarenal extracranial rhabdoid tumor (EERT) are seen in children with a reported incidence of 0.15/million in children less than 15 years old and it is rarer in adults.[7] Children with EERT tend to be older and present in less advanced stage, compared with those with RTK and commonly have involvement of the musculoskeletal system.[8]
This tumor poses considerable diagnostic dilemma, and radiologically, it may mimic malignant peripheral nerve sheath tumor (MPNST) in the paraspinal region, the other differentials being neurofibroma, schwannoma, ganglioneuroma, paraganglioma, and meningioma.[2] Histopathologically, it may be mistaken for a variety of “blue cell” sarcomas and other tumors, for example, ewing sarcoma, rhabdomyosarcoma, synovial sarcoma, MPNST, lymphoma, and melanoma.[2] At a molecular level, the entity is defined by mutation or alteration in the SMARB1/INI1 gene resulting in loss of INI1 expression, which is pathognomonic of rhabdoid tumor.[1,2,7]
The aggressive clinical course of this tumor is analogous to that of RTK. In a series of 106 children diagnosed with extracranial MRT in the UK from 1993 to 2010, 1-year survival was only 31%.[9] In a single-institution series of 14 cases of extracranial MRT diagnosed over a 20-year period, the median time to progression was only 2 months.[6] Horazdovsky et al. demonstrated improved survival with older age at diagnosis (>2 years) in patients with EERT.[10]
EERT being extremely rare, there is no definite treatment guideline, and an aggressive multimodality approach is recommended.[9] Radical surgical resection is the treatment of choice and was associated with 74% improvement in overall survival (OS) in a meta-analysis of 167 cases of EERT.[10] In a study of 21 cases of the extracranial rhabdoid tumor by Venkatramani et al., all patients with unresectable primary tumor died at the last follow-up.[11] However, even after complete resection, local recurrence is frequent. Cai et al. described a case of adult pelvic retroperitoneal ERRT, in which tumor progressed locally 2 weeks after surgery and metastasized to lungs, abdominal wall, and mesenteric lymph nodes.[12]
The active chemotherapeutic drugs in MRT are alkylating agents (cyclophosphamide/ifosfamide), anthracycline (doxorubicin/epirubicin), actinomycin D, platinum (cisplatin/carboplatin), epipodophyllotoxin (etoposide), and vinca alkaloid (vincristine). There is no randomized controlled trial examining the role of chemotherapy in patients with EERT in view of the rarity of the tumor. In a meta-analysis of 167 cases of EERT, use of actinomycin D led to 73% improvement in OS.[10] European Paediatric Soft Tissue Sarcoma Study Group (EpSSG) conducted a multinational single arm prospective study from 2005 to 2012 to test an intensive multimodal treatment approach for children with newly diagnosed extracranial MRT.[9] After initial surgery or biopsy, intensive multiagent chemotherapy was administered over 30 weeks – vincristine, cyclophosphamide, and doxorubicin at weeks 1, 10, 13, 22, and 28; cyclophosphamide, carboplatin, and etoposide at weeks 4, 7, 16, 19, and 25 and single agent vincristine at all other weeks. With this approach, the event-free survival was 32.3% and OS was 38.4% at 3 years. The inclusion of doxorubicin in chemotherapy regimen is suggested to be important for optimal survival in patients with extracranial MRT.[13]
Radiotherapy is believed to be an essential component of multimodality treatment in patients with MRT. In a case series of three patients with EERT treated by surgery, radiotherapy, and chemotherapy by Puri et al., two patients survived and were disease-free at 6.5 and 7 years after diagnosis.[14] In a SEER report of 229 patients with MRT diagnosed from 1986 to 2005, age at diagnosis (2–18 years), localized stage at presentation and use of radiotherapy significantly improved OS.[8] However, lack of data, pertaining to dose, and volume of radiotherapy, was a limitation of this report. Radiotherapy was recommended for all sites of primary and metastatic disease at week 2 after upfront surgery or week 14 after delayed surgery in the EpSSG study.[9] The dose of radiotherapy depended on the site of primary and metastatic disease and the broad guidelines were 36 Gray, 45 Gray, and 50.4 Gray (at 1.8 Gray per fraction per day) after R0, R1, and R2 resection, respectively, in patients with EERT.
To sum up, MRT is a rare entity with a dismal prognosis. The most common sites of involvement are kidney and brain, and pelvis is a relatively rare site. This tumor has been commonly described in the pediatric age group and presentation in adulthood is quite rare. We have described a case of MRT involving the pelvic bones with soft tissue component in a young male adult. Initially, this case was misdiagnosed as bone Tuberculosis with tubercular abscess at a local center and use of ATT led to local disease progression as well as pulmonary metastasis. This underscores the importance of awareness of various bone and soft tissue tumors of the pelvis, including MRT, in a young adult, in a country where Tuberculosis is rampant. In view of metastatic disease, the illustrative patient underwent 6 cycles of chemotherapy with ICE regimen. In spite of partial response at the local site, there was an eventual progression in the form of liver metastasis. Radiotherapy was considered to the primary site and led to worthwhile palliation of local symptoms. Thereafter, the patient was started on metronomic chemotherapy which led to prolonged disease stabilization.
Conclusion
EERT is a rare disease with a grave prognosis. Despite aggressive therapy, it is difficult to achieve long-term cure in patients with metastatic disease. Symptomatic improvement and prolonged disease stabilization can be achieved by a judicious combination of systemic chemotherapy and radiotherapy to primary and metastatic sites. Metronomic chemotherapy may be considered in patients with progressive or refractory disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Molina N, Leis A. Extrarenal rhabdoid tumor of the brachial plexus in a five-year-old female: A case report and review of the literature. J Pediatr Surg Case Rep 2016;15:5-9.
- Dobbs MD, Correa H, Schwartz HS, Kan JH. Extrarenal rhabdoid tumor mimicking a sacral peripheral nerve sheath tumor. Skeletal Radiol 2011;40:1363-8.
- Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol 1989;13:439-58.
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms' tumor: Results of the First National Wilms' Tumor Study. Cancer 1978;41:1937-48.
- Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol 1981;12:646-57.
- Madigan CE, Armenian SH, Malogolowkin MH, Mascarenhas L. Extracranial malignant rhabdoid tumors in childhood: The childrens hospital Los Angeles experience. Cancer 2007;110:2061-6.
- Raspollini MR, Li Marzi V, Nicita G, Mikuz G. The challenging diagnosis of the rhabdoid carcinoma of the pelvis: A case report with literature review. Appl Immunohistochem Mol Morphol 2012;20:177-83.
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, Nassan AA, Ghandour K, Al-Hussaini M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 2010;54:35-40.
- Brennan B, De Salvo GL, Orbach D, De Paoli A, Kelsey A, Mudry P, et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer 2016;60:69-82.
- Horazdovsky R, Manivel JC, Cheng EY. Surgery and actinomycin improve survival in malignant rhabdoid tumor. Sarcoma 2013;2013:315170.
- Venkatramani R, Shoureshi P, Malvar J, Zhou S, Mascarenhas L. High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer 2014;61:1357-61.
- Cai G, Zhu X, Xu Y, Du X, Zhang Z, Zhang Y, et al. Case report of extrarenal rhabdoid tumor of pelvic retroperitoneum molecular profile of angiogenesis and its implication in new treatment strategy. Cancer Biol Ther 2009;8:417-21.
- Waldron PE, Rodgers BM, Kelly MD, Womer RB. Successful treatment of a patient with stage IV rhabdoid tumor of the kidney: Case report and review. J Pediatr Hematol Oncol 1999;21:53-7.
- Puri DR, Meyers PA, Kraus DH, Laquaglia MP, Wexler LH, Wolden SL. Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 2008;50:167-9.
| Figure 1:(a) Malignant tumor composed of pleomorphic cells and rhabdoid cells with vesicular nuclei, prominent nucleoli and intracytoplasmic eosinophilic inclusion (arrow). Immunohistochemistry shows (b) diffuse strong expression of cytokeratin, (c) vimentin and (d) loss of expression of INI-1 in nuclei of tumor cells
| Figure 2:(a and b) Computed tomography scan of pelvis with oral and intravenous contrast showing a lesion arising from the left side of sacrum with involvement of left sacro-iliac joint and left iliac bone, extradural extension through S1–S2 sacral foramina, intrapelvic soft tissue extension, infiltration of the left psoas muscle, and extension into the left groin; (c) computed tomography scan of thorax-lung window showing a suspicious nodule in lower lobe of right lung suggestive of metastasis; (d) contrast-enhanced computed tomography scan of abdomen showing an ill-defined lesion in segment 4a of liver suggestive of metastasis
References
- Molina N, Leis A. Extrarenal rhabdoid tumor of the brachial plexus in a five-year-old female: A case report and review of the literature. J Pediatr Surg Case Rep 2016;15:5-9.
- Dobbs MD, Correa H, Schwartz HS, Kan JH. Extrarenal rhabdoid tumor mimicking a sacral peripheral nerve sheath tumor. Skeletal Radiol 2011;40:1363-8.
- Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol 1989;13:439-58.
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms' tumor: Results of the First National Wilms' Tumor Study. Cancer 1978;41:1937-48.
- Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol 1981;12:646-57.
- Madigan CE, Armenian SH, Malogolowkin MH, Mascarenhas L. Extracranial malignant rhabdoid tumors in childhood: The childrens hospital Los Angeles experience. Cancer 2007;110:2061-6.
- Raspollini MR, Li Marzi V, Nicita G, Mikuz G. The challenging diagnosis of the rhabdoid carcinoma of the pelvis: A case report with literature review. Appl Immunohistochem Mol Morphol 2012;20:177-83.
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, Nassan AA, Ghandour K, Al-Hussaini M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 2010;54:35-40.
- Brennan B, De Salvo GL, Orbach D, De Paoli A, Kelsey A, Mudry P, et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer 2016;60:69-82.
- Horazdovsky R, Manivel JC, Cheng EY. Surgery and actinomycin improve survival in malignant rhabdoid tumor. Sarcoma 2013;2013:315170.
- Venkatramani R, Shoureshi P, Malvar J, Zhou S, Mascarenhas L. High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer 2014;61:1357-61.
- Cai G, Zhu X, Xu Y, Du X, Zhang Z, Zhang Y, et al. Case report of extrarenal rhabdoid tumor of pelvic retroperitoneum molecular profile of angiogenesis and its implication in new treatment strategy. Cancer Biol Ther 2009;8:417-21.
- Waldron PE, Rodgers BM, Kelly MD, Womer RB. Successful treatment of a patient with stage IV rhabdoid tumor of the kidney: Case report and review. J Pediatr Hematol Oncol 1999;21:53-7.
- Puri DR, Meyers PA, Kraus DH, Laquaglia MP, Wexler LH, Wolden SL. Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 2008;50:167-9.