 PDF
PDF  Views
Views  Share
Share


Emergence of micronuclei as a genomic biomarker
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2015; 36(04): 212-218
DOI: DOI: 10.4103/0971-5851.171541
Abstract
The presence of micronuclei (MN) in mammalian cells is related to several mutagenetic stresses. MN are formed as a result of chromosome damage and can be readily identified in exfoliated epithelial cells. MN is chromatin particles derived from acentric chromosomal fragments, which are not incorporated into the daughter nucleus after mitosis. It can be visualized by chromatin stains. A variety of factors influences the formation of MN in cells such as age, sex, genetic constitution, physical and chemical agents, adverse habits such as tobacco, areca nut chewing, smoking, and alcohol consumption. Micronucleation has important implications in the genomic plasticity of tumor cells. The present paper reviews the origin, fate and scoring criteria of MN that serves as a biomarker of exposure to genetic toxins, and for the risk of cancer.
Publication History
Article published online:
12 July 2021
© 2015. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
The presence of micronuclei (MN) in mammalian cells is related to several mutagenetic stresses. MN are formed as a result of chromosome damage and can be readily identified in exfoliated epithelial cells. MN is chromatin particles derived from acentric chromosomal fragments, which are not incorporated into the daughter nucleus after mitosis. It can be visualized by chromatin stains. A variety of factors influences the formation of MN in cells such as age, sex, genetic constitution, physical and chemical agents, adverse habits such as tobacco, areca nut chewing, smoking, and alcohol consumption. Micronucleation has important implications in the genomic plasticity of tumor cells. The present paper reviews the origin, fate and scoring criteria of MN that serves as a biomarker of exposure to genetic toxins, and for the risk of cancer.
INTRODUCTION
There is an increasing effort worldwide to determine the impact of environmental, genetic and life-style factors on genomic stability in human populations. One technique that has been adopted by numerous laboratories is the measurement of micronuclei (MN) in peripheral blood lymphocytes, epithelial cells, erythrocytes and fibroblasts.[1]
Micronuclei were first described by Howell and Jolly in late 1800's and early 1900's as Feulgen positive nuclear bodies in human reticulocytes, representing chromosomes separated from the mitotic spindle.[2]
Micronuclei are characteristically seen in exfoliated epithelial cells like buccal mucosa and urinary bladder wall during precancerous and cancerous conditions.[3]
The buccal cell micronucleus is defined as the microscopically visible, round or oval cytoplasmic chromatin mass next to nucleus. MN originate from aberrant mitosis and consist of acentric chromosomes, chromatid fragments or whole chromosomes that have failed to be incorporated in the daughter nuclei during mitosis.[4]
These are also seen in various conditions like chronic tonsillitis,[5] chronic renal diseases[6] and rheumatoid arthritis.[7]
Tobacco is used socially in the form of cigarettes, biddies, cigars where it is smoked, and it is also chewed in various forms like gutka, pan masala, khaini. The effect of tobacco on the oral tissues varies depending upon the type, frequency and duration of use.
Tobacco-specific nitrosamines have been reported to be potent clastogenic and mutagenic agents that are thought to be responsible for the induction of chromatid/chromosomal aberrations resulting in the production of MN. The genotoxic and carcinogenic chemicals released from betel nut and tobacco and also the calcium hydroxide content of lime present in the betel quid are thought to be responsible for the promotion of reactive oxygen species from areca nut extracts. These reactive oxygen species can in turn cause damage to the deoxyribonucleic acid (DNA).[8]
The MN count is increased in potentially malignant disorders like oral sub mucous fibrosis, leukoplakia, erythroplakia, lichen planus and squamous cell carcinoma.[9,10]
Thus, the quantitative estimation of MN may serve as an indicator of genetic damage that has taken place. Oral carcinogenesis is a multi-step process of accumulated genetic damage leading to cell dysregulation with disruption in cell signaling. These events can be conveniently studied in the buccal mucosa, which is an easily accessible tissue for sampling cells in a minimally invasive manner.[11]
The buccal mucosa provides a barrier to potential carcinogens that can be metabolized to generate potential reactive products. Exfoliated buccal cells have been used noninvasively to successfully show the genotoxic effects of lifestyle factors such as tobacco smoking, chewing of betel nuts and/or quids, medical treatments, such as radiotherapy as well as occupational exposure to potentially mutagenic and/or carcinogenic chemicals, and for studies of chemoprevennstion of cancer.[12]
The present goal in many research laboratories is to develop screening strategies indicating individual cancers with certain biomarkers. Biomarkers are instruments of individual tumor prevention and help to detect high-risk patients. Biomarkers are divided into three groups: The first to define the exposure to carcinogenic agents, the second to show biological effects on the target tissue and the third to give information about the individual susceptibility.[11]
Scoring of the MN can be performed relatively easily and on different cell types relevant for human biomonitoring: Lymphocytes, fibroblasts and exfoliated epithelial cells.[1]
MICRONUCLEI FORMATION
Chromosomal aberrations are a frequent and significant response to exposure to mutagenic agents. They are of significance from the standpoint of inherited human disease and have been in carcinogenesis.[13]
Micronuclei are one of such biomarkers that are cytoplasmic chromatin masses with the appearance of small nuclei that arise from lagging chromosomes at anaphase or from acentric chromosome fragments [Figure 1]. These are formed by chromosomal damage in the basal cells of the epithelium. When these cells divide, chromosomal fragments (or entire chromosomes that lack attachment to the spindle apparatus) lag behind and are excluded from the main nuclei in the daughter cells [Figure 2]. These fragments form their own membranes and are termed micronucleus in the cell cytoplasm, and these cells later mature and are exfoliated.[11]
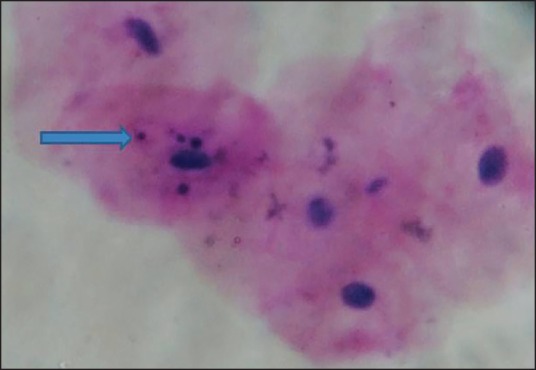
| Fig. 1 Oral exfoliated cells showing micronuclei in oral mucosa (×40)

| Fig. 2 Micronucleus expression in a dividing cell[1]
Such MN are induced by genotoxic stress such as clastogens or aneugens. The agents that cause chromosomal breaks are called clastogens (radiation); and which act on the spindle are called aneuploidogens or aneugens (vincristine).[14]
Micronuclei induction by clastogen involves the induction of either chromosome fragments that lag behind the separating chromosomes or a chromatin bridge between chromosomes at the anaphase of mitosis. On the other hand, aneugen induce the whole chromosomes that are not bound to the mitotic spindle at anaphase, probably by disrupting the spindle checkpoint. Such chromatin is separated from the newly forming nucleus and forms an independent nucleus-like structure, the micronucleus.[15]
Most MN appear directly after completion of mitosis, although some appeared long after mitosis is completed. Early MN are derived from either the chromatin bridge between separating anaphase chromosomes or the chromatid detached from the bulk of the chromosomes during the transition from metaphase to anaphase. The detached chromatid located between the separating anaphase chromosomes may be merotelically attached to the spindle. Unexpectedly, detached chromatids can also be located at position closer to the spindle pole than the chromosomes or at the side of the chromosomes, possibly reflecting syntactical or monotelical attachment to the spindle. Chromatids located in these positions produced MN.
Part of the chromatin bridge might remain as the nuclear bud (NBUD) after its resolution. This constitutes for 29% of the buds that are detected just after mitosis. On the other hand, the buds might also appear at the end of mitosis in the absence of detectable chromatin bridges. This type of bud constituted 28% of the total buds. Furthermore, the buds generated long after the apparently normal mitosis, typically by the protrusion of the interphase nucleus constitute 43% of all buds. Some of the NBUDs may be converted to MN. Therefore, methods to measure the frequency of MN can widely be used as genotoxic test to measure efficacy of newly developed pharmaceuticals or used to diagnose malignant disease.[16]
THE FATE OF MICRONUCLEATED CELLS
The frequency of apoptotic cell death is much higher among the cells bearing MN compared with cells bearing normal nuclei. If the MN-bearing cells enter mitosis, they either produced daughter cells without MN or, more frequently, produce cells with additional MN.[16]
SCORING CRITERIA
Criteria for identifying and scoring cell types in exfoliated cell micronucleus assay
Buccal cells can be categorized into normal cells and the cells that are considered abnormal on the basis of cytological and nuclear features. These include:[12]
- Normal basal cells.
- Normal differentiated cells.
- Cells with MN.
- Cells with NBUDs.
- Binucleated cells.
- Buccal cells with condensed chromatin.
- Karyorrhectic cells.
- Pyknotic cells.
- Karyolytic cells.
- Normal basal cells have a larger nucleus-to-cytoplasmic ratio than the differentiated buccal cells. Basal cells have a uniformly stained nucleus and are smaller in size and more oval in shape when compared to the more angular and flat differentiated buccal cells. No DNA-containing structures apart from the nucleus are observed in these cells. The cytoplasm is typically stained a darker shade of green with light green compared to the differentiated cells.
- Normal “differentiated” cells have a uniformly stained nucleus, which is oval or round in shape. They are distinguished from basal cells by their larger size and by a smaller nucleus-to cytoplasmic ratio. No other DNA-containing structures apart from the nucleus are observed in these cells.
- Cells with MN are characterized by the presence of both a main nucleus and one or more smaller nuclear structures called MN. The MN is round or oval in shape, and their diameter should range between 1/3 and 1/16 of the main nucleus. MN has the same staining intensity and texture as the main nucleus. Most cells with MN will contain only one MN but it is possible to find cells with two or more MN. Baseline frequencies for micronucleated cells in the bone marrow are usually within the 0.5-2.5 mni/1,000 cells range.
- Cells with nuclear buds contain nuclei with an apparent sharp constriction at one end of the nucleus suggestive of a budding process, that is the elimination of nuclear material by budding. The NBUD and the nucleus be usually in very close proximity and appear to be attached to each other. The NBUD has the same morphology and staining properties as the nucleus; however, its diameter may range from a half to a quarter of that of the main nucleus. The mechanism leading to NBUD formation is not known but it may be related to the elimination of amplified DNA or DNA repair.
- Binucleated cells are cells containing two main nuclei instead of one. The nuclei are usually very close and may touch each other and usually have the same morphology as that observed in normal cells. The significance of these cells is unknown, but they are probably indicative of failed cytokinesis following the last nuclear division in the basal cell layer.
- Buccal cells with condensed chromatin show a roughly striated nuclear pattern in which the aggregated chromatin is intensely stained. These cells may be undergoing early stages of apoptosis.
- Karyorrhectic cells have nuclei that are characterized by more extensive nuclear chromatin aggregation relative to condensed chromatin cells. They have a densely speckled nuclear pattern indicative of nuclear fragmentation leading to the eventual disintegration of the nucleus. These cells may be undergoing a late stage of apoptosis.
- Pyknotic cells are characterized by a small shrunken nucleus, with a high density of nuclear material that is uniformly but intensely stained. The nuclear diameter is usually one- to two-thirds of a nucleus in normal differentiated cells. The biological significance of the pyknotic cells represents an alternative mechanism of nuclear disintegration that is distinct from the process leading to the condensed chromatin and karyorrhectic cell death stages.[17]
- Karyolytic cells are cells in which the nucleus is completely depleted of DNA and is apparent as a ghost-like image that has no Feulgen staining. Therefore, these cells appear to have no nucleus and represent a very late stage in the cell death process [Figure 3].
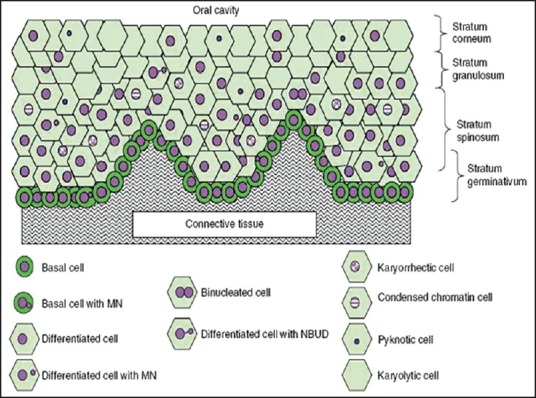
| Fig. 3 Diagrammatic representation of a cross section of normal buccal mucosa. The mucosa of healthy individuals illustrating the different cell layers and possible spatial relationships of the various cell types are shown [Nature Protocols 2009]
Criteria for the inclusion in total cell count
Heddle initially described the well-established basic criteria for MN.[18] However, the criteria for identifying cells for inclusion into the MN frequency count were not provided. Later Tolbert et al. developed the criteria for choosing the cells, and this is being most widely applied.[19,20] It consists of the following parameters:
- Cytoplasm intact and lying relatively flat.
- Little or no overlap with adjacent cells.
- Little or no debris.
- Nucleus normal and intact, nuclear perimeter smooth and distinct.
In order for a cell to be considered micronucleated the cell must satisfy the above criteria regarding inclusion in total cell count and the suggested criteria for identifying MN are:
- Rounded, smooth perimeter suggestive of membrane.
- Less than a third the diameter of the associated nucleus but large enough to discern shape and color.
- Feulgen positive (i.e., pink in bright field illumination).
- Staining intensity is similar to that of the nucleus.
- Texture similar to that of the nucleus.
- Same focal plane as nucleus.
- Absence of overlap or bridge to nucleus.
Tolbert et al. also recommended the scoring of at least 1000 cells, with an increase to 2000-3000 if fewer than 5 micronucleated cells were observed after counting 1000 cells. The majority of the published studies have scored between 1000 and 3000 cells, although it has been suggested that 10,000 cells may be needed to observe a statistically significant, 50% increase, in the MN frequency.[20,21]
FACTORS AFFECTING THE SCORING OF MICRONUCLEI
A few studies reported the possibility that cellular structures, such as keratohyalin granules or bacteria, resembling MN, can lead to false-positive results. Keratohyalin granules are reported to occur in cells of the granular layer of interfollicular epidermis of the skin, and are predominantly composed of the 400-kDa protein profillagrin. MN scoring can be interfered by the bacteria that are commonly found in the mouth. Bacteria can be differentiated from MN by their characteristic shape, smaller size, color, staining intensity, and their presence upon and between buccal cells on the slide. Small dye granules may sometimes resemble MN but usually have a slightly different refractility and color intensity.[22]
HUMN MICRONUCLEUS PROJECT
The International Collaborative project on Micronucleus frequency in human populations (HUMN) was organized to collect data on micronucleus frequencies in different human populations and different cell types. The test procedures considered by this project are assays using human lymphocytes (cytokinesis-block method), exfoliated epithelial cells and other cell types. Data including descriptions of the populations monitored detailed test protocols, and test results are being obtained from a large number of laboratories throughout the world and are being entered into a unified database. The information will be used to:[23]
- Determine the extent of variation of ‘normal’ values for different laboratories and the influence of other factors potentially affecting baseline MN frequency, e.g., age, gender and life-style.
- Provide information on the effect of experimental protocol variations on MN frequency measurements.
- Design and test optimal protocols for the different cell types.
- Determine the extent to which MN frequency is a valid biomarker of ageing and risk for diseases such as cancer.
CYTOMETRIC ANALYSIS OF MICRONUCLEI
Flow cytometry
Flow cytometry was originally used to study distinct cell populations within a mixture of different cell types. With this technique, a fluorescent dye is used to specifically label the cell population of interest. Individual cells can then be examined one at a time as they are pulled through the flow cytometer and subjected to laser-diffracted light to determine cell size and shape. Fluorescently labeled cells can also be sorted into separate tubes, based on their size and the intensity of their fluorescence signal, using diffraction plates in a process called fluorescence-activated cell sorting.[24]
The majority of the flow cytometric work was based on the approach of Nusse and Kramer that involved lysis of outer membranes. In conjuction with one or more nucleic acid dyes, it was possible to discriminate the liberated nuclei and MN based on their DNA-dye associated fluorescence intensities.[25,26]
While flow cytometry was clearly shown to be a high throughput platform with great potential, the early methods experienced problems discriminating MN from cellular debris in the cell populations, Later modifications to those of Nusse and Kramer attempted to overcome these problems, taking advantage of the higher resolution of later machines and using improved gating based on a combination of light scatter and fluorescence signals to distinguish MN signals from debris.
The reliability of flow cytometry-based MN scoring was improved when the procedures of Nusse et al. were modified and subsequently configured into a commercially available kit. The most significant of these modifications were (i) incorporation of a fluorescent dye to differentiate MN from chromatin associated with dead and dying cells and (ii) incorporation of concurrent assessments of cell/culture health that identify overly cyotoxic treatment conditions that tend to lead to unreliable MN measurements.
The current state of the art utilizes a dual dye sequential staining procedure. More specifically at the time of harvest, intact cells are incubated with ethidium monoazide bromide (EMA). electrophoretic mobility shift is a nucleic acid that enters the compromised membranes of necrotic and late stage apoptotic cells. A unique feature of EMA is that it can be covalently bound to DNA following a photoactivation step. This makes it possible to label dead/dying cells with EMA without loss of signals as cells are further processed. This EMA labeling step is followed by exposure to a detergent-containing lysis solution that includes the nucleic acid dye SYTOX green, which provides pan-DNA labeling. In this manner, the sequential dye protocol results in differential staining of healthy cells chromatin (EMA−/SYTOX+) relative to necrotic and late stage apoptotic cells (EMA+/SYTOX+) profile. By excluding EMA+ chromatin from analysis, flow cytometric scoring of nuclei and MN is accomplished with significantly reduced interference from the presence of dead/dying cells.[27]
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) involves the use of fluorescently labeled DNA probes that are capable of hybridizing to complementary chromosomal regions. This technique allows researchers to view the chromosomal location of a particular gene or DNA sequence through a microscope; the net result is a fluorescent dot at the chromosomal location where the labeled probe binds. FISH can also be used to identify genes with increased copy number or to detect gene loss, as shown by more or fewer than two fluorescent “dots” in a somatic cell, respectively. Furthermore, FISH can be carried out using nondividing cells, which allows investigators to examine nonmitotic cells. This is important, because DNA packing is approximately 10,000 times less compact in nonmitotic (interphase) cells, allowing researchers to achieve a higher level of resolution. An extremely high-resolution form of FISH, called fiber-FISH, is carried out using isolated chromosomes that are free from nuclear architecture and exist as long, stretched-out DNA fibers.[24]
Micronuclei arises from the chromosomal fragments or whole chromosomes that are left behind during mitotic division. The dual origin of MN can be distinguished using FISH with probes labeling the centromeric region of all human chromosomes, so MN derived from acentric chromosomal fragments will not be labeled by the probe and the MN harboring whole chromosomes will be positively labeled. This approach was first introduced by Becker and co-workers in human lymphocytes and then applied to buccal cells. Thus, the FISH/MN assay potentially allows a quick and reliable identification of both aneugenic and clastogenic effects in buccal cells. The MN assay has the potential limitation that it only detects missegregating chromosomal fragments or whole chromosomes, but is unable to detectstable chromosome aberrations such as translocations. This limitation can be overcome by using chromosome 1tandem labeling FISH. Tandem labeling FISH is performed by using α satellite, biotin labeled DNA probe that hybridizes specifically with the centromeric region of chromosome 1and a classical satellite, digoxigenin labeled DNA probe detecting the adjacent heterochromatic band 1q12.[28]
Spectral karyotyping and multiplex-fluorescence in situ hybridization
The ability to isolate individual human chromosomes using flow cytometry, combined with knowledge of the human genome sequence, has allowed cytogeneticists to develop 24-color probe sets that are used to label each human chromosome with a distinct color. Chromosome specific probes are made by labeling DNA fragments covering the length of each individual chromosome with a distinctly colored fluorescent dye. The labeled DNA probes are then pooled and used in hybridization experiments with metaphase chromosome spreads. The labeled DNA probe sets bind to their complementary chromosomes, allowing each individual chromosome to be labeled with a specific fluorescent color along its entire length. In a somatic cell, the maternal and paternal copy of each chromosome will be labeled with the same colors. This powerful approach, which permits the simultaneous tracking of all human chromosomes, has been called spectral karyotyping (SKY) or multiplex-FISH (M-FISH).[24]
Limitations of micronucleus test
The MN assay can be influenced by changes in the mitotic rate or the proportion of cell death. To avoid a misinterpretion of MN frequency estimates in exfoliated cells, few unresolved methodological issues must be mentioned.
- Depending on the tissue, the migration of cells from the basal layer to the epithelium may require a few days or a few weeks. During this time span, micronucleated cells could conceivably lyse and disappear from the epithelium before reaching the surface layers. Thus, on analyzing exfoliated surface cells, the actual frequency of micronucleated cells may be underestimated.
- A second source of error could be introduced by an incorrect sampling time. The frequency of MN in the surface cells will reflect events which took place several days before in the basal layer of dividing cells. Moreover, the period between the induction of chromosome aberrations in the basal layer and the appearance of MN in exfoliated cells can be delayed or enhanced by the action of carcinogens.
- Any carcinogenic effect which does not involve a damage to the chromosome complement will not be reflected by elevated frequencies of micronucleated cells.
- If a tissue is only occasionally hit by a carcinogen, then a single sample of exfoliated cells could fail to detect a temporary increase in micronucleated cells. In such cases, repeated sampling would be the method of choice.
- Different laboratories have employed different criteria for MN scoring, making inter laboratory comparisons difficult.[19]
CONCLUSION
Micronuclei are the extra nuclear cytoplasmic bodies. They are induced in oral exfoliated cells by a number of substances, including genotoxic agents and carcinogenic compounds in tobacco, betel nut and alcohol that damage the chromosomes. The damaged chromosomes in the form of accentric chromatid or chromosomes fragments lag behind in anaphase, when the centric elements move towards the spindle poles. After telophase the undamaged chromosomes and the centric fragments give rise to regular daughter nuclei. The lagging elements are included in the daughter cells too, but a considerable portion is transformed into one or several secondary nuclei, which are as a rule much smaller than the main nucleus and are therefore called as MN. The analysis of MN in exfoliated mucosal cells can be used as a biomarker of genotoxicity in predicting the effects of carcinogens.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Oral exfoliated cells showing micronuclei in oral mucosa (×40)
| Fig. 2 Micronucleus expression in a dividing cell[1]
| Fig. 3 Diagrammatic representation of a cross section of normal buccal mucosa. The mucosa of healthy individuals illustrating the different cell layers and possible spatial relationships of the various cell types are shown [Nature Protocols 2009]