 PDF
PDF  Views
Views  Share
Share


Embryonal carcinoma in androgen insensitivity syndrome
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(02): 105-108
DOI: DOI: 10.4103/0971-5851.89794
Abstract
Embryonal cell carcinoma is a rare clinical entity. We report a case of a 20-year-old patient who presented with lump lower abdomen for last two months with primary amenorrhea and poorly developed secondary sexual characteristics. Ultrasonography (USG) whole abdomen showed lower abdominal mass approximately 15΄15΄10 cm, probably neoplastic changes in intra-abdominal testis, with mild ascites, no uterus and ovaries. Fine needle aspiration cytology from the tumor mass reported the possibility of non-seminomatous germ cell tumor, possibly embryonal carcinoma. The patient received three cycles of neo-adjuvant chemotherapy (Regime Bleomycin, Etoposide and Cisplatin) followed by laparotomy, at laparotomy (L) orchidectomy with removal of tumor, (R) orchidectomy, omentectomy and appendisectomy was performed. Postoperatively the patient received two more cycles of chemotherapy of the same regime. The patient has been under close follow-up for the last three years with no evidence of disease.
Publication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Embryonal cell carcinoma is a rare clinical entity. We report a case of a 20-year-old patient who presented with lump lower abdomen for last two months with primary amenorrhea and poorly developed secondary sexual characteristics. Ultrasonography (USG) whole abdomen showed lower abdominal mass approximately 15×15×10 cm, probably neoplastic changes in intra-abdominal testis, with mild ascites, no uterus and ovaries. Fine needle aspiration cytology from the tumor mass reported the possibility of non-seminomatous germ cell tumor, possibly embryonal carcinoma. The patient received three cycles of neo-adjuvant chemotherapy (Regime Bleomycin, Etoposide and Cisplatin) followed by laparotomy, at laparotomy (L) orchidectomy with removal of tumor, (R) orchidectomy, omentectomy and appendisectomy was performed. Postoperatively the patient received two more cycles of chemotherapy of the same regime. The patient has been under close follow-up for the last three years with no evidence of disease.
INTRODUCTION
Androgen insensitivity syndrome (AIS), also called testicular feminization syndrome, is a form of X-linked male pseudohermaphroditism that is clinically recognized in patients with a female phenotype. The incidence of this syndrome has been reported in 1: 20,400 newborns.
CASE REPORT
Miss AB, a 20-year-old female, attended our outpatients’ department (OPD) on 16 August 2007 with complaints of lump lower abdomen for last two months [Figure 1]. She also complained of nausea and vomiting for the last few days. She had history of primary amenorrhea and poorly developed secondary sexual characteristics. There was no history of cyclic abdominal pain, hormonal intake, radiation exposure or any chemotherapy. There was no history of significant trauma and medical or surgical illness. There was no history of any major disease in the family. She is the second child of her mother being delivered vaginally at the age of 29 years. The mother reported that the pediatrician after examining the baby told the parents that the external genitalia of the baby were inadequately developed and advised the mother to return after one year if there was no adequate development. Over time they did not turn up and she started growing up as a female although with very poor development of her secondary sex characters. She has one brother and one sister. All of them attained puberty normally and are enjoying good health.
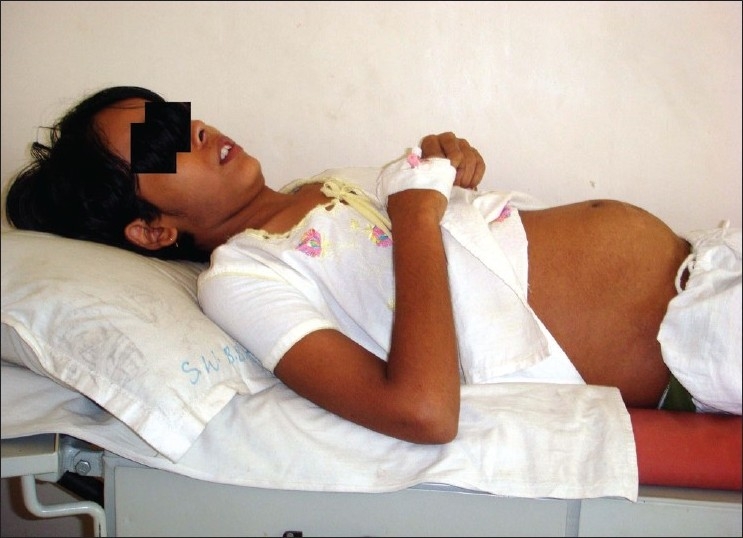
| Fig. 1 Preoperative clinical presentation
On general examination her condition was fair, height 146 cm and weight 42 kg. She had mild pallor. There was no cyanosis, clubbing, edema, obesity, acne, hirsutism, thyroid swelling or cushingoid features; her breast was small, underdeveloped with hypopigmented areola (Tanner Stage II). There was no axillary or pubic hair. Her systemic examination including chest and Cardiovascular system were within normal limits. Per abdomen examination revealed lump in lower abdomen, there was no visible swelling in the inguinal region. Her external genitalia were infantile. There were hypertrophied clitoris resembling like a micropenis. She had inadequately developed vulva with small vaginal pit [Figure 2].

| Fig. 2 External genitalia
Her blood group was A +ve, H aemoglobin percentage 7.5 g% and all other routine blood tests were within normal limits. Her S. AFP was 48815.2 IU/ml (normal range < 7.22 IU/ml), serum beta hCG=1.24 mIU/ml (normal range < 6.15 mIU/ml), serum prolactin and thyroid profile were within normal limits, serum testosterone level 924 ng/dl (normal range 300-1000 ng/dl), serum estradiol 31.18 pg/ml, serum calcium 7.6 mg% and Parathyroid hormone level 9.69 pg/ml. Karyotyping showed 46 xy pattern. A whole abdomen ultrasonography (USG) showed no uterus and ovaries. There was a lower abdominal mass approx 15 cm × 15 cm × 10 cm, probably neoplastic changes in an intra-abdominal testis, with mild ascites.
A USG-guided fine needle aspiration cytology (FNAC) from the tumor mass reported the possibility of non-seminomatous germ cell tumor, possibly embryonal carcinoma. Smear from ascitic fluid was suggestive of metastatic adenocarcinoma. Computed tomography (CT) scan of brain revealed normal findings. PA view chest X-ray was normal.
A diagnosis of Stage IIIC malignant germ cell tumor (embryonal carcinoma) of undescended (intra-abdominal) gonad was made. The tumor board of our Institute decided to go in for laparotomy following a few courses of neoadjuvant chemotherapy.
She received three cycles of neoadjuvant chemotherapy (Regime Bleomycin, Etoposide and Cisplatin). Post chemotherapy an assessment of the disease status was done by a CT scan whole abdomen, which revealed an oval-shaped residual solid mass arising from the left side of the pelvis and externally to the lower abdominal cavity; the size of the mass was 12 × 7.2 cm × 9.5 cm. No adjacent organ infiltration was noted. No ascitis, vascular or ureteric encasement was seen. No hepatic secondary or significant lymphadenopathy was noted. Serum AFP came down to 3.4 IU/ml and S. B. hCG: 3.24 mIU/ml. After proper counseling and written and informed consent, laparotomy was done on 10 December 2007 under general anesthesia. The findings were: left testis was enlarged, 15×10 cm, right testis was of normal size, no uterus/ovaries detected.
At laparotomy (L) orchidectomy with removal of tumor, (R) orchidectomy, omentectomy and appendisectomy was performed [Figures [Figures33–5]. Histopathological examination of the excised specimens: Left testis: embryonal cell carcinoma, Right testis– normal, omentum – metastatic deposit, appendix- chronic appendicitis. Her postoperative recovery was uneventful. She was discharged on 18– December 2007 and advised to attend the Gynecology OPD after three weeks at which her S AFP level was 2.3 IU/ml.
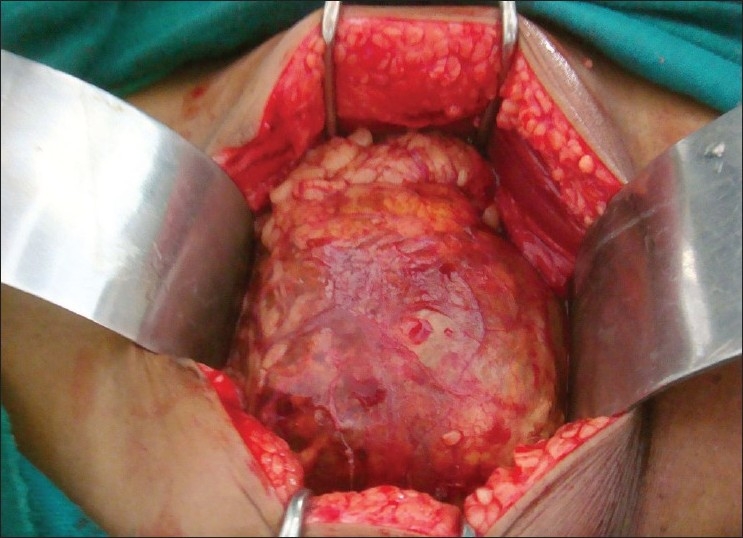
| Fig. 3 Left testicular tumor, intraoperative
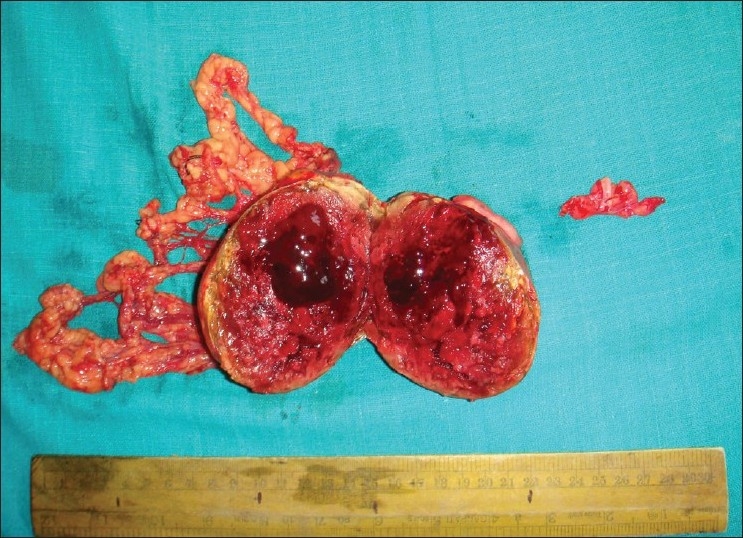
| Fig. 5 Postoperative specimen of left testicular tumor cut open
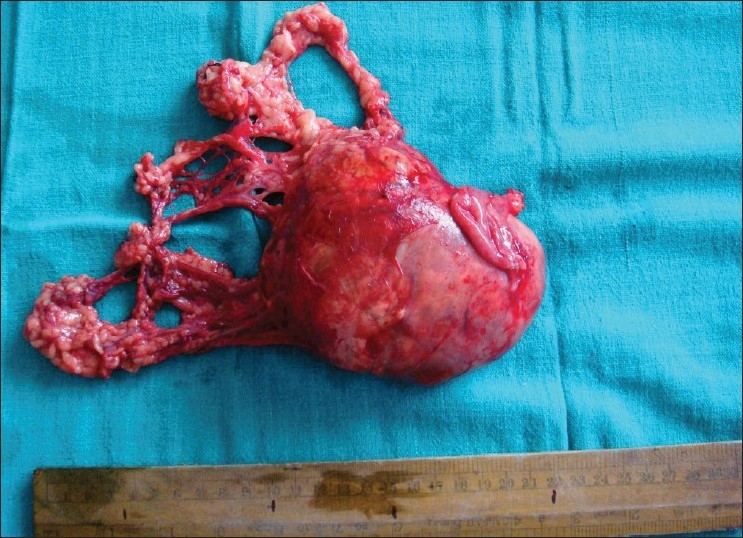
| Fig. 4 Postoperative specimen of left testicular tumor
Postoperatively she received two more cycles of adjuvant chemotherapy of the same regime.
On 25 February 2008 repeat total S. testosterone was 120 ng/dl and free testosterone 940 ng/dl.
Over four months after operation she was doing well and her secondary sexual characters were developing (breast development Tanner Stage III) after taking hormonal supplementation advised by the endocrinologist. She is on regular follow-up, she attended our OPD for checkup on 20 October 2009 with no evidence of disease clinically, USG was within normal limits and normal S. AFP.
DISCUSSION
In androgen insensitivity syndrome (AIS), there is failure of normal masculinisation of the external genitalia in chromosomally male individuals. This failure of virilisation can be either complete androgen insensitivity syndrome (CAIS), or partial androgen insensitivity syndrome (PAIS), depending on the amount of residual receptor function. In post-pubertal patients, the most frequent cause for consultation is primary amenorrhea; however, in pre-pubertal patients, inguinal hernia may be the only symptom. The diagnosis of CAIS /male pseudohermaphroditism (46, XY) can be difficult in the neonate period, sometimes the only clue to suggest the diagnosis is an inguinal hernia in these females, who display a normal phenotype. Newborn girls with inguinal hernias have a 1.6% incidence of being male pseudohermaphrodites (46, XY) and should undergo a prompt karyotype analysis.
The karyotype is 46 XY, although cases with chromosomal mosaicism have also been reported.[1] Individuals with CAIS have female external genitalia with normal labia, clitoris and vaginal introitus. The phenotype of individuals with PAIS may range from mildly virilized female external genitalia, clitorimegaly without other external anomalies to mildly undervirilized male external genitalia (hypospadias and/or diminished penile size).[2–4]
The basic etiology of AIS is a loss of function mutation in the androgen receptor (AR) gene. This AR gene has been localized to the long arm of the X chromosome (i.e. Xq11-13). Over 200 such mutations have been described, including complete and partial gene deletions, point mutations, and small insertions/deletions.[5]
In patients with CAIS, the testes should be removed after pubertal development is complete to prevent malignant transformation. In patients with cryptorchidism, the risk of developing germ cell tumor is increased fourfold to eightfold. The risk is 1% if the testis is retained in the inguinal canal. The risk of developing germ cell tumor when a cryptorchid testis is intra-abdominal is about 5%. Almost half of the testicular neoplasms are malignant (dysgerminoma), but transformation usually does not occur until after puberty. In patients who develop virilisation and have an XY karyotype, the testes should be removed immediately to preserve the female phenotype and female gender identity. Bilateral laparoscopic gonadectomy is the preferred procedure for removal of intra-abdominal testes.[6–7]
The histology in our case was typical of non-seminomatous germ cell tumor embryonal carcinoma, which mostly occurs in the 20-30 years age group. These tumors are more aggressive than seminomas. Although pure embryonal cell carcinoma constitutes about 3% of testicular germ cell tumor, if one includes the mixed tumors, then embryonal carcinoma cells is present in about 45% of testicular germ cell tumors.[8]
Our patient had omental metastasis which is very rare with few reported cases but at the same time it is difficult to explain if it is due to seedling or due to prior FNAC.[9] Management of non-seminomatous germ cell tumor Stage III constitutes neoadjuvant chemotherapy with BEP regime followed by surgery and the overall cure rate is around 50–70%, depending on risk factors.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 Preoperative clinical presentation
| Fig. 2 External genitalia
| Fig. 3 Left testicular tumor, intraoperative
| Fig. 5 Postoperative specimen of left testicular tumor cut open
| Fig. 4 Postoperative specimen of left testicular tumor