 PDF
PDF  Views
Views  Share
Share


Cutaneous Infarcts and Antineutrophil Cytoplasmic Antibody?Positive Vasculitis in Angioimmunoblastic T?Cell Lymphoma: A Rare Presentation
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2019; 40(04): 598-600
DOI: DOI: 10.4103/ijmpo.ijmpo_133_18
Sir,
Angioimmunoblastic T-cell lymphoma (AITL) is a rare and unique subtype of peripheral T-cell lymphoma, accounting for 2%–5% of all non-Hodgkin’s lymphomas.[1] The lymph node architecture is often effaced, with retention of only a few benign follicles. The infiltrate characteristically extends beyond the lymph node capsule, with preserved and dilated subcapsular sinus. Often, there is interfollicular proliferation of endothelial venules. The neoplastic T-cells are positive for CD2, CD3, CD4, CD10, CXCL-13, PD1, and often BCL-6.[2]
AITL is associated with many systemic features such as fever, rash, or arthritis. These systemic features may precede the appearance of other features of lymphoma. The disease may present as autoimmune phenomena, such as the presence of circulating immune complexes, cold agglutinins, hemolytic anemia, and rheumatoid factor and anti-smooth muscle antibodies. Hypergammaglobulinemia is present in approximately 50% of patients, typically polyclonal.[3] Vasculitis is occasionally associated with AITL, but it is usually antineutrophil cytoplasmic antibody (ANCA)-negative vasculitis.[4]
Publication History
Received: 14 June 2018
Accepted: 12 July 2019
Article published online:
03 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Sir,
Angioimmunoblastic T-cell lymphoma (AITL) is a rare and unique subtype of peripheral T-cell lymphoma, accounting for 2%–5% of all non-Hodgkin’s lymphomas.[1] The lymph node architecture is often effaced, with retention of only a few benign follicles. The infiltrate characteristically extends beyond the lymph node capsule, with preserved and dilated subcapsular sinus. Often, there is interfollicular proliferation of endothelial venules. The neoplastic T-cells are positive for CD2, CD3, CD4, CD10, CXCL-13, PD1, and often BCL-6.[2]
AITL is associated with many systemic features such as fever, rash, or arthritis. These systemic features may precede the appearance of other features of lymphoma. The disease may present as autoimmune phenomena, such as the presence of circulating immune complexes, cold agglutinins, hemolytic anemia, and rheumatoid factor and anti-smooth muscle antibodies. Hypergammaglobulinemia is present in approximately 50% of patients, typically polyclonal.[3] Vasculitis is occasionally associated with AITL, but it is usually antineutrophil cytoplasmic antibody (ANCA)-negative vasculitis.[4]
Case
A 65-year-old male, a nonsmoker with no known comorbidities, presented with complaints of short febrile illness associated with weight loss, dry cough, generalized weakness, pruritus, and paresthesia in the bilateral lower limbs. He was admitted to the medical ward, 12 h after which the patient developed dyspnea, blackish discoloration of digits [Figure 1], and cutaneous necrotic lesions over the bilateral legs [Figure 2]. Clinical examination revealed scattered polyphonic wheeze bilaterally over the chest. A working diagnosis of secondary vasculitis was made, and the patient was started on methylprednisolone pulse in view of impending digital gangrene.

| Fig. 1 Digital infarcts in a 65-year-old male patient, who is a nonsmoker with no known comorbidities and presented with complaints of short febrile illness associated with weight loss, dry cough, generalized weakness, pruritus, and paresthesia in the bilateral lower limbs
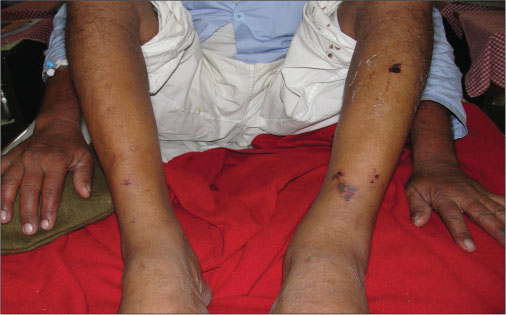
| Fig. 2 Cutaneous necrotic lesions over the bilateral legs in the same patient. A working diagnosis of secondary vasculitis was made, and the patient was started on methylprednisolone pulse in view of impending digital gangrene
He responded to the treatment with no further progression of digital ischemia. Evaluation revealed anemia (Hb = 5–6 g/dl), thrombocytopenia (platelet = 5000/mm3), raised erythrocyte sedimentation rate (ESR) (78 mm/h), and eosinophilia (AEC-2910). Biochemistry panel revealed azotemia (serum urea/serum creatinine = 70/3.5 mg/dl). Autoimmune workup revealed ANCA positive (pANCA) strongly by ELISA; however, antinuclear antibody and direct and indirect Coombs test were negative. Tumor markers including AFP, CEA, and CA-19-9 were negative. Contrast-enhanced computed tomography chest and abdomen showed hilar and pretracheal lymphadenopathy and hepatosplenomegaly, respectively [Figure 3] and [Figure 4]. Magnetic resonance angiography of the aorta and its branches showed small, scarred right kidney with small caliber of the right renal artery, but there was no evidence of aortoarteritis. Nerve conduction studies were suggestive of mononeuritis multiplex with the involvement of ulnar and peroneal nerves.
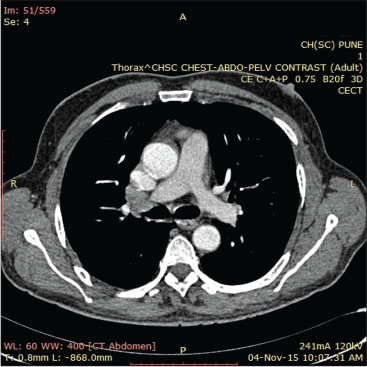
| Fig. 3 Contrast-enhanced computed tomography chest showing mediastinal lymphadenopathy
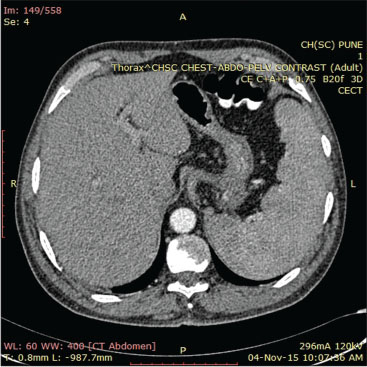
| Fig. 4 Contrast-enhanced computed tomography abdomen showing hepatosplenomegaly
Bone marrow studies showed cellular (80%) and reactive marrow picture with slight increase in the plasma cells. Serum protein electrophoresis revealed monoclonal gammopathy (immunoglobulin G lambda restricted). A lymph node biopsy of the left axilla was done, and histopathological examination was suggestive of AITL [Figure 5], [Figure 6], [Figure 7]. He was started on chemotherapy (CHOP regimen) and has shown response.
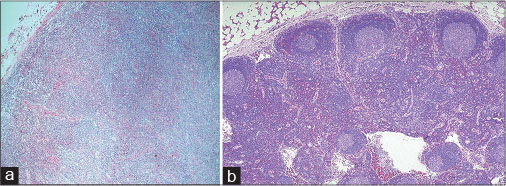
| Fig. 5 (a and b) Hematoxylin and eosin stain on a lymph node biopsy sample (from left axilla) showing effaced lymph node architecture
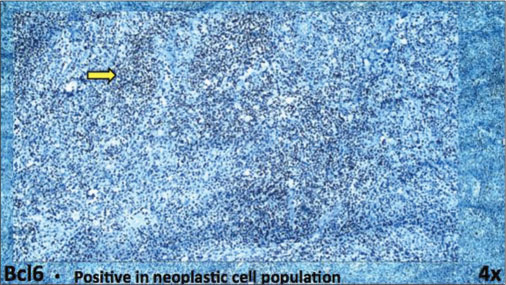
| Fig. 6 Immunohistochemistry on a lymph node biopsy sample (from left axilla) showing BCL6 positivity

| Fig. 7 Immunohistochemistry on a lymph node biopsy sample (from left axilla) showing CD3 and CD5 positivity
Discussion
AITL is a rare and aggressive neoplasm clinically manifesting as sudden onset of constitutional symptoms, lymphadenopathy, hepatosplenomegaly, frequent autoimmune phenomena, particularly hemolytic anemia and thrombocytopenia, and polyclonal hypergammaglobulinemia.[5]
The median age of the patients is 60–65 years, with an equal incidence in males and females. In most of the cases, the clinical spectrum of the disease includes B-symptoms (70%), pruritus/skin rash (50%), hepatomegaly (50%), splenomegaly (79%), pleural effusion (37%), edema (40%), and ascites (25%).[3] In a significant proportion of patients, autoimmune diseases can be seen, including autoimmune hemolytic anemia, cold agglutinin anemia, vasculitis, polyarthritis, and autoimmune thyroiditis. Among laboratory investigations, the most frequent findings on diagnosis are anemia, eosinophilia, hypergammaglobulinemia, autoantibodies, elevated lactate dehydrogenase serum levels, elevated ESR, and bone marrow involvement.
In general, paraneoplastic vasculitis associated with lymphoid malignancies is ANCA negative; however, our patient presented with vasculitis, which was pANCA. There are two reported cases of pANCA vasculitis, one with AITL and other with chronic lymphocytic leukemia, with both patients being cANCA positive.[4]
AITL is associated with hypergammaglobulinemia, but rarely, monoclonal gammopathy has been reported. Our patient had monoclonal gammopathy with features simulating POEMS syndrome with neuropathy, organomegaly, skin changes, and endocrine dysfunction.
Conclusion
This case emphasizes the atypical presentation of a rather rare disease and the need for high index of suspicion and close follow-up for the diagnosis of AITL. The early diagnosis will help in initiating chemotherapy well in time, which will help prolong median survival rates in patients suffering from this aggressive disease.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- 1 Rüdiger T, Weisenburger DD, Anderson JR, Armitage JO, Diebold J, MacLennan KA. et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): Results from the non-Hodgkin’s lymphoma classification project. Ann Oncol 2002; 13: 140-9
- 2 Attygalle AD, Diss TC, Munson P, Isaacson PG, Du MQ, Dogan A. CD10 expression in extranodal dissemination of angioimmunoblastic T-cell lymphoma. Am J Surg Pathol 2004; 28: 54-61
- 3 Frizzera G, Moran EM, Rappaport H. Angio-immunoblastic lymphadenopathy with dysproteinaemia. Lancet 1974; 1: 1070-3
- 4 Hamidou MA, El Kouri D, Audrain M, Grolleau JY. Systemic antineutrophil cytoplasmic antibody vasculitis associated with lymphoid neoplasia. Ann Rheum Dis 2001; 60: 293-5
- 5 Iannitto E, Ferreri AJ, Minardi V, Tripodo C, Kreipe HH. Angioimmunoblastic T-cell lymphoma. Crit Rev Oncol Hematol 2008; 68: 264-71
Address for correspondence
Dr. Sandeep KumarDepartment of General Medicine, Army College of Medical SciencesDelhi Cantt, New Delhi - 110 010IndiaEmail: sandeep.sapehia@gmail.comPublication History
Received: 14 June 2018
Accepted: 12 July 2019
Article published online:
03 June 2021© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig. 1 Digital infarcts in a 65-year-old male patient, who is a nonsmoker with no known comorbidities and presented with complaints of short febrile illness associated with weight loss, dry cough, generalized weakness, pruritus, and paresthesia in the bilateral lower limbs
| Fig. 2 Cutaneous necrotic lesions over the bilateral legs in the same patient. A working diagnosis of secondary vasculitis was made, and the patient was started on methylprednisolone pulse in view of impending digital gangrene
| Fig. 3 Contrast-enhanced computed tomography chest showing mediastinal lymphadenopathy
| Fig. 4 Contrast-enhanced computed tomography abdomen showing hepatosplenomegaly
| Fig. 5 (a and b) Hematoxylin and eosin stain on a lymph node biopsy sample (from left axilla) showing effaced lymph node architecture
| Fig. 6 Immunohistochemistry on a lymph node biopsy sample (from left axilla) showing BCL6 positivity
| Fig. 7 Immunohistochemistry on a lymph node biopsy sample (from left axilla) showing CD3 and CD5 positivity
References
- 1 Rüdiger T, Weisenburger DD, Anderson JR, Armitage JO, Diebold J, MacLennan KA. et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): Results from the non-Hodgkin’s lymphoma classification project. Ann Oncol 2002; 13: 140-9
- 2 Attygalle AD, Diss TC, Munson P, Isaacson PG, Du MQ, Dogan A. CD10 expression in extranodal dissemination of angioimmunoblastic T-cell lymphoma. Am J Surg Pathol 2004; 28: 54-61
- 3 Frizzera G, Moran EM, Rappaport H. Angio-immunoblastic lymphadenopathy with dysproteinaemia. Lancet 1974; 1: 1070-3
- 4 Hamidou MA, El Kouri D, Audrain M, Grolleau JY. Systemic antineutrophil cytoplasmic antibody vasculitis associated with lymphoid neoplasia. Ann Rheum Dis 2001; 60: 293-5
- 5 Iannitto E, Ferreri AJ, Minardi V, Tripodo C, Kreipe HH. Angioimmunoblastic T-cell lymphoma. Crit Rev Oncol Hematol 2008; 68: 264-71