 PDF
PDF  Views
Views  Share
Share


Current therapy and recent advances in the management of retinoblastoma
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2012; 33(02): 80-88
DOI: DOI: 10.4103/0971-5851.99731
Abstract
Retinoblastoma is the most common intraocular malignancy in children. The survival of retinoblastoma patients and visual outcome has improved dramatically in the developed world. This can be attributed to early tumor recognition and advances in the management of retinoblastoma. Chemoreduction followed by adjuvant consolidative treatment has replaced external beam radiotherapy as the primary modality of treatment for intraocular retinoblastoma. Further, histopathological high-risk factors have been identified in enucleated eyes, allowing use of prophylactic chemotherapy to take care of possible micrometastasis. The survival in case of extraocular retinoblastoma is still low, and the reported survival rate ranges between 50% and 70%. In developing countries, the overall survival of retinoblastoma patients remains low, primarily due to a delayed presentation, resulting in larger proportions of extraocular disease compared with the developed world, where majority of the disease is intraocular. Greater efforts need to be directed toward early tumor recognition in order to improve the survival of retinoblastoma patients in the developing world. In this article, we provide an overview of the current clinical management of retinoblastoma.
Publication History
Article published online:
13 April 2022
© 2012. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Retinoblastoma is the most common intraocular malignancy in children. The survival of retinoblastoma patients and visual outcome has improved dramatically in the developed world. This can be attributed to early tumor recognition and advances in the management of retinoblastoma. Chemoreduction followed by adjuvant consolidative treatment has replaced external beam radiotherapy as the primary modality of treatment for intraocular retinoblastoma. Further, histopathological high-risk factors have been identified in enucleated eyes, allowing use of prophylactic chemotherapy to take care of possible micrometastasis. The survival in case of extraocular retinoblastoma is still low, and the reported survival rate ranges between 50% and 70%. In developing countries, the overall survival of retinoblastoma patients remains low, primarily due to a delayed presentation, resulting in larger proportions of extraocular disease compared with the developed world, where majority of the disease is intraocular. Greater efforts need to be directed toward early tumor recognition in order to improve the survival of retinoblastoma patients in the developing world. In this article, we provide an overview of the current clinical management of retinoblastoma.
INTRODUCTION
Retinoblastoma is the most common intraocular malignancy in children, and is reported to affect 1 in 15,000 to 1 in 18,000 live births.[1] It represents almost 4% of all pediatric malignancies. Approximately 250–300 children are newly diagnosed with retinoblastoma each year in the United States, and substantially higher rates occur in developing countries.[2,3] Studies from India show a two- to three-fold higher incidence of tumors of the eye (majority of which will be retinoblastoma in children < 15 years of age).[4]
Also, while over 95% children with retinoblastoma survive in the developed world, only 50% children with retinoblastoma survive worldwide.[3] This can be explained by a delay in presentation and, consequently, a greater number of cases with extraocular invasion seen in developing countries.
Extraocular retinoblastoma is very rare in developed countries (reported incidence: approximately 2–5%).[5] In developing nations, extraocular disease contributes to half of all retinoblastoma cases presenting to a tertiary care referral center.[6] A combination of poverty, illiteracy, alternative systems of medicine and lack of access to healthcare resources account for this high rate of advanced disease in developing countries.
In this article, we will review the current state of art for managing retinoblastoma. We searched the PubMed for important articles on recent advances in the treatment of retinoblastoma over the last 2 decades, and reviewed them with special reference to the state of retinoblastoma in India.
DIAGNOSIS AND INVESTIGATIONS
Clinical profile
Retinoblastoma generally presents any time from birth to 5 years of age, although in rare cases it may present later. In the United States, the mean age at presentation is 18 months. There is no sex predilection. Retinoblastoma is unilateral in approximately two-thirds of cases and bilateral in the remaining one-third.
The most common presentation is with cat's eye reflection in the pupil. Other less common clinical presentations include a uveitis-like picture, aseptic orbital cellulitis, phthisis bulbi, hyphema, buphthalmos, red, painful eye with glaucoma and cloudy cornea, proptosis and even fungating mass. Patients with metastasis may present with bone pains, vomiting and headache or scalp masses in addition. Family history may be present in less than 10% of the cases.
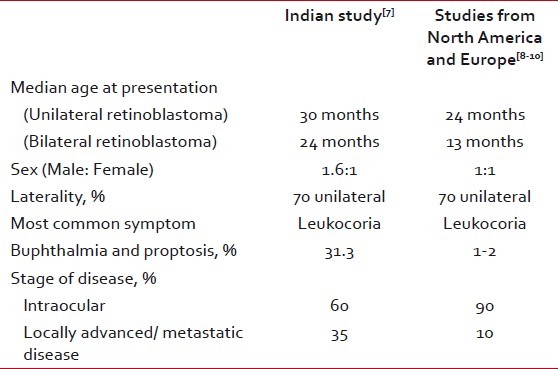
In a study conducted at the All India Institute of Medical Sciences, the median age of presentation of retinoblastoma patients was 2.5 years (median age for unilateral retinoblastoma was 3 years and for bilateral retinoblastoma was 2 years). Sixty-nine percent of the cases were unilateral and 30.5% were bilateral. There was a male predominance, with a male to female ratio of 1.6:1. The most common first symptom noted by parents was white reflex, followed by redness or squint. Proptosis was the most common symptom (31.3%) at presentation apart from white reflex. The median duration of symptoms before presentation was 7.2 months. Sixty percent of the patients presented with intraocular disease, 32.72% with locally advanced disease and 10.16% with metastatic disease [Table 1].[7]
Table 1
Clinical features of retinoblastoma
Clinical evaluation
All children with suspected retinoblastoma undergo a preliminary clinical evaluation in the clinic, at initial visit, consisting of visual acuity assessment, the pupillary examination, slit-lamp examination and indirect ophthalmoscopy. Subsequently, all patients undergo examination under anesthesia (EUA), consisting of a intraocular pressure measurement using a portable tonometer, corneal diameters, slit-lamp examination for evidence of iris neovascularization, hyphema or anterior segment involvement in the form of iris nodules or hypopyon; binocular indirect ophthalmoscopy with 360 degrees of scleral depression; and fundus photographs (RETCAM) documenting all lesions. If RETCAM is not available, handmade fundus diagrams documenting the site, size and numbers of retinal tumors, vitreous and subretinal seeds and retinal detachment are to be drawn at each EUA.
Imaging
The diagnosis of retinoblastoma is generally made on indirect ophthalmoscopy. Imaging studies are used for (1) confirmation of diagnosis in difficult cases, (2) when there is opaque media precluding indirect ophthalmoscopy and (3) to evaluate the presence of extraocular extension or associated cerebral lesion in trilateral cases.
The imaging diagnosis of retinoblastoma is based on documentation of an intraocular mass with calcification. If an ultrasound is available in the outpatient department/clinic, mass with calcification may be picked up in most cases. However, if it is not available or calcification is not picked up on ultrasound, a computed tomography (CT) scan is recommended for documenting the same.
Although magnetic resonance imaging (MRI) is more sensitive than CT scan for picking up extraocular extension, CT scan is more widely used in developing countries because of the ease of availability and affordability. Although a CT scan may detect extraocular extension in a majority of the cases, some cases are likely to be missed out if MRI is not done routinely.
MRI will still be indicated in cases where there is a doubt of extraocular extension on CT scan. MRI has the advantage of distinguishing tumor with secondary retinal detachment from other causes of subretinal effusion or detachment, like coat's disease, primary hyperplastic vitreous and retinopathy of prematurity.[11] Also, some authors believe that performing CT scan in cases with bilateral retinoblastoma may increase the risk of secondary malignancies later in survivors; MRI does not have any such risk.[12] Currently, MRI is the standard imaging modality used to rule out extraocular extension.
Once the diagnosis is established, the disease is staged based on the clinical and imaging findings, and intraocular retinoblastoma is classified in order to determine appropriate line of treatment.
Classification of intraocular retinoblastoma
The Reese-Ellsworth classification was developed in the era where radiotherapy was the mainstay for treating retinoblastoma. With the emergence of chemoreduction as the core of therapy for intraocular retinoblastoma, the variables affecting the outcome of treatment have changed. Vitreous and subretinal seeds have been identified as the major cause of treatment failure or relapse. The recently developed International Classification for Intraocular Retinoblastoma[13] takes into account the factors that affect the outcome (globe salvage and survival) in the present treatment scenario [Table 2].
Table 2
International classification of intraocular retinoblastoma

Staging of retinoblastoma
Retinoblastoma if not treated on time has the ability to metastasize to the central nervous system via the optic nerve, bone and bone marrow through blood and lymph nodes through lymphatics. Staging is based on the systemic extent of the tumor and, hence, prognosticates survival. Various ocular oncologists have used different staging systems.
The recently introduced International Retinoblastoma Staging System (IRSS) is based on histopathology and imaging findings, and takes into account the observation that extraocular retinoblastoma with spread to regional lymph nodes has a rate of overall survival that is comparable to nonmetastatic extraocular retinoblastoma[14] [Table 3].
Table 3
International retinoblastoma staging system

Metastatic work-up
Initial metastatic work-up consists of lumbar punctures for cerebrospinal fluid analysis (CSF), bone marrow evaluation and bone scan to exclude brain, bone marrow and bone metastases, respectively. In an analysis of 259 patients with retinoblastoma, Bakhshi et al. reported that none of the stage 0 or I cases tested positive on CSF or bone marrow evaluation despite the presence of histopathologic high-risk factors. They concluded that there is no role of CSF and bone marrow evaluation in stage 0 and I retinoblastoma; these tests must be done in stage III and IV patients to rule out microscopic metastasis. However, stage II patients need to be evaluated further.[15]
Screening for retinoblastoma
Parents and siblings of the affected child should be screened for evidence of retinoblastoma in all cases of retinoblastoma in order to identify the hereditary cases of retinoblastoma.
TREATMENT
The primary aim of therapy is to save the patients’ life. Globe salvage and saving vision in the affected eye are also important but secondary aims of treatment. A treatment plan is formulated in collaboration with the ophthalmologist, pediatric oncologist and pediatric radiation oncologist [Table 4]. There are three possible modalities of treatment:
Table 4
Indications for various treatment modalities in retinoblastoma

- Surgery
- Chemotherapy
- Radiotherapy
Surgical options
Enucleation
Primary enucleation is the choice of treatment for unilateral group E and D intraocular retinoblastoma. Cases of intraocular retinoblastoma that have failed on chemotherapy and conservative treatment are also treated with enucleation. Further, it is now possible to carry out enucleation in most cases of overt orbital disease (stage III) following two to three cycles of neoadjuvant chemotherapy (NACT) [Table 4].
Exenteration
It is an extremely disfiguring surgery. Upfront exenteration is now obsolete. It is currently performed only in those cases of primary/recurrent orbital disease that fail to respond to NACT.
Focal consolidative measures
These are used in early intraocular tumors (ICRB A–D) following chemoreduction (two to three cycles) or primarily, and include the following options:
- a)Laser photocoagulation: It is employed for tumors not more than 2 mm in height and located posterior to equator.
- b)Thermotherapy: This is a method of tumor heating, where the aim is to achieve a temperature of 42–60 degrees, which is below the coagulative threshold. It is used for tumors not larger than 3 mm in basal dimensions located at the posterior pole or mid-periphery and without any vitreous seeding. It has a synergistic effect with chemotherapy and may be used in conjunction with chemotherapy for large tumors.
- c)Cryotherapy: It is used to treat equatorial or peripheral small tumors that do not exceed 3 mm in diameter and 1.5 mm in thickness.
- d)Chemothermotherapy: All tumors situated at the posterior pole or in the mid-peripheral retina, with a diameter of 15 mm or more without vitreous seeding and not suitable for treatment by cryoapplication or thermotherapy alone, may be treated using a combination of transpupillary thermotherapy and chemotherapy.
CHEMOTHERAPY
The chemotherapeutic agents most commonly used in the management of retinoblastoma include vincristine, etoposide and carboplatin. Other drugs that may be used include cyclophosphamide, melphalan and thiotepa.
Chemotherapy may be used in the following four possible ways in treating retinoblastoma [Table 4]:
Chemoreduction
The aim of chemoreduction is to reduce the tumor volume in large intraocular tumors (ICRB Group B to D), thereby allowing use of less-damaging and more-focused therapeutic measures in order to preserve vision and avoid enucleation. The most common chemotherapy protocol used currently consists of vincristine, etoposide and carboplatin [Table 4]. Adequate tumor reduction requires two to six cycles of chemotherapy. Following two cycles of chemotherapy, Shields et al. have reported a 35% decrease in the mean base diameter, 50% decrease in the tumor thickness and complete resolution of sub-retinal fluid in 76% cases.[16,17,18] They have also reported a 25% recurrence of subretinal seeds when less than six cycles were administered compared with 0% recurrence in cases who received six cycles of chemotherapy.[1] Therefore, it is believed that less than six cycles of chemoreduction may not be sufficient to completely destroy sub-retinal seeds. Also, focal treatment is essential to prevent tumor recurrence. Wilson et al. reported a tumor recurrence rate of 92% in 36 eyes with retinoblastoma that were treated with eight cycles of chemotherapy (vincristine and carboplatin) alone, without any tumor consolidation.[19] Shields et al. reported a tumor recurrence in 45% cases at 7 years of follow-up in cases treated with chemotherapy alone compared with only 22% recurrence in those where tumor consolidation was done following chemoreduction using focal treatment measures.[20]
Periocular chemotherapy
The major problem with systemic chemotherapy is the recurrence of vitreous and sub-retinal seeds.[21] It is believed that this may occur because of limited penetration of systemically administered chemotherapeutic agents into the sub-retinal and vitreous spaces, which are avascular. Use of local delivery techniques (periocular) allows higher effective doses at these sites, establishing effective treatment while limiting the systemic side-effects of chemotherapy. Most centers use carboplatin in an aqueous vehicle (same as used for systemic therapy). Although periocular carboplatin has been reported to have promising therapeutic benefit in the treatment of groups C and D retinoblastoma, it is associated with serious adverse effects, including ocular motility changes, orbital fat necrosis, severe pseudopreseptal cellulitis and ischemic necrosis with atrophy of the optic nerve resulting in blindness.[22–24] These side-effects are thought to result from a rapid dispersal of the chemotherapeutic agent. Therefore, clinical trials are on to assess the effects of periocular carboplatin delivered in a fibrin sealant that allows for a controlled-release of the drug in the periocular space.[25,26] Topotecan in fibrin sealant has also been tried subconjunctivally to treat retinoblastoma successfully in the mouse model of retinoblastoma.[27]
Intrarterial chemotherapy
Recently, intraarterial injection of the chemotherapy drug melphalan in retinoblastoma patients has been tried. A group of Japanese researchers have been investigating intracarotid administration of chemotherapy.[28,29] However, in their studies, many eyes also received external beam radiotherapy (EBRT) and intravitreal melphalan, making it difficult to determine how effective the intraarterial injections would be if used alone. Data from a phase I/II clinical trial recently conducted by Abramson and associates has demonstrated good outcomes after direct intraophthalmic artery injections of melphalan. They used selective catheterization of the ophthalmic artery, thereby theoretically avoiding toxicity to the brain. Vision stabilized or improved in all but one of the nine patients enrolled in the study. The side-effect profile reported was benign.[30] Many patients, however, also received supplementary thermotherapy, laser, brachytherapy or EBRT, making it difficult to determine whether this method is effective in increasing the globe salvage rates on its own. Despite its remarkable control for retinoblastoma, particularly recurrent tumor seeds following other therapies, some authors feel that intraarterial chemotherapy should be used with caution.[31] Recently, Shields et al. have evaluated the toxic effects of irradiation from fluoroscopy during intraarterial chemotherapy for retinoblastoma, and have cautioned that accumulated irradiation toxic effects following multiple sessions of intraarterial chemotherapy could be cataractogenic and possibly carcinogenic.[32]
Chemoprophylaxis
Several studies have shown that presence of certain histopathological features in the enucleated eyes is associated with a higher risk of systemic metastasis and/or local recurrence.[33–35] Adjuvant treatment in these cases reduces the likelihood of local recurrence and distant metastasis.[36,37] Histopathological features, which are used as an indication for six cycles of adjuvant chemotherapy at most centers, include invasion of anterior chamber, iris, ciliary body, choroid (massive), sclera and optic nerve beyond lamina cribrosa. Additionally, extrascleral and optic nerve cut-end involvement is classified as microscopic residual disease and is treated with 12 cycles of adjuvant chemotherapy and EBRT.
Chemotherapy for locally advanced disease
NACT is used to reduce the tumor size, followed by enucleation, local radiotherapy and adjuvant chemotherapy, to complete a total of 12 cycles. NACT leads to rapid reduction in tumor bulk and also controls micrometastatic disease. The reduction in tumor bulk enables most patients to undergo enucleation instead of cosmetically disfiguring and more complex exenteration.
Most centers currently use standard doses of vincristine, carboplatin and etoposide (VEC) for neoadjuvant and adjuvant chemotherapy. However, data pertaining to use of high-dose versus standard dose of carboplatin is lacking in the absence of any randomized control trials comparing the same. The overall survival reported in patients with extraocular disease is 50–70% in patients with extraocular retinoblastoma[38] Data from India shows an overall survival of 50% at 18 months of follow-up.[39] The role of intracranial prophylaxis with intrathecal chemotherapy or cranial irradiation in patients with locally advanced retinoblastoma has not been prospectively evaluated. Outcomes in retrospective studies with or without intrathecal chemotherapy have been similar. Because of lack of evidence, the use of intrathecal chemotherapy as prophylaxis to prevent central nervous system metastases in locally advanced retinoblastoma remains experimental.[40]
High-dose chemotherapy and autologous stem cell transplant
Autologous stem cell transplant with high-dose chemotherapy remains the only curative option for metastatic disease. There have been only few case reports/series in the literature with regards to the use of this modality for the treatment of metastatic disease.[41] Children with noncentral nervous system disease (bone or bone marrow) have had better outcomes when compared with patients with central nervous system metastasis. Long-term survival of 50–75% has been reported in patients with noncentral nervous system disease in a few series.[42] The drugs commonly used as part of high-dose chemotherapy include combinations of carboplatin, etoposide, cyclophosphamide, melphalan and thiotepa. The role of allogenic transplant remains investigational.[43]
RADIOTHERAPY
Retinoblastoma is a radiosensitive tumor and radiotherapy is used in various stages of retinoblastoma.
For intraocular tumors, radiotherapy may be delivered as [Table 4]:
Plaque therapy (brachytherapy)
It is indicated in cases of peripheral tumors up to 15 mm in diameter and 7–8-mm thick, possibly with localized vitreous invasion over the tumor.
External beam radiotherapy
EBRT has mostly been replaced by aggressive chemoreduction and focal consolidative therapy for intraocular disease. But, it still remains useful in cases of very large or multiple tumors, unsuitable for other focal treatment modalities (thermotherapy, cryoapplication, radioactive plaque or chemothermotherapy) and after failure of other techniques. A dose of 45 Gray is delivered to the target volume by two electron beams over 5 weeks, with fractions of 1.8 (for children under 12 months old) to 2 Gy (for children over 12 months).
However, radiation damage to the ocular structures can be challenging to manage. Also, it may induce secondary cancers in the field of radiation, with a reported 30-year cumulative incidence for second cancers in bilateral retinoblastoma treated with irradiation at 35% compared with 6% in those who did not receive radiation.[44] Further, the risk is greater for patients who are younger than 1 year of age at the time of treatment.[45] In a recent study on effectiveness of EBRT as a salvage treatment after failed primary chemotherapy and focal treatment in bilateral retinoblastoma, the authors found salvage EBRT to be highly effective in preserving eyes with useful vision in bilateral retinoblastoma after failed chemotherapy and focal treatments. Sixty-six percent of tumors were controlled by EBRT alone and required no further treatment and 52.6% had a visual acuity of 6/9 or better in the salvaged eye.[46]
EBRT in extraocular retinoblastoma
Local EBRT is an important component of the treatment protocol for eradication of residual disease in the orbit. The usual dose of radiotherapy given to the involved orbit is 40–45 Gray over 4–5 weeks. There is no role for prophylactic cranial irradiation. The inclusion of optic chiasma in the radiation field is not clear and is practiced by few centers.[47]
The molecular signaling pathways in retinoblastoma and potential clinical applications
The normal retinoblastoma protein binds to and inhibits the transcription factor E2F, thereby halting transcription of E2F target genes, which are responsible for cell cycle progression. It also binds to histone deacetylase (HDAC), which results in silencing of transcription. Mutated or absent retinoblastoma protein results in uncontrolled cellular proliferation and, ultimately, cancer. Little progress has been made toward the development of targeted therapeutics to benefit retinoblastoma. The potential targets for molecular therapy include:
Nutlin 3A
Nutlin 3A is a small molecule inhibitor of the MDM2/MDMX and p53 interaction, which was found to be able to induce cell death in human retinoblastoma cell lines.[48] Further, when used in combination with topotecan, it killed human retinoblastoma cell lines. There was an 82-fold reduction in tumor burden in mice when Nutlin 3A was injected subconjunctivally with topotecan.[49]
Inhibitors of HDAC
HDAC inhibitors may be particularly useful because they induce cytotoxicity to tumors selectively.[50] Recently, it was found that certain HDAC inhibitors reduce cell survival in human retinoblastoma cell lines and significantly reduced tumor burden in both rat and mouse models of retinoblastoma.[51]
Molecule inhibitors of the oncogene N-myc
N-myc is amplified in 10% of human retinoblastoma cases and, therefore, such inhibitors may prove useful in the treatment of a subset of retinoblastoma patients.[52]
CHALLENGES IN TREATING RETINOBLASTOMA IN OUR COUNTRY
As India is a resource-challenged country, cost constraints typical to such a setup that make availability of ideal modalities for imaging, like MRI and photo documentation with RETCAM, difficult. The main challenge in treating retinoblastoma effectively in our country is a delay in presentation, resulting in advanced disease and consequently higher treatment-related morbidity and disease-related mortality. Further, the compliance to treatment is also poor.
The compliance rate in retinoblastoma patients in a study from AIIMS was found to be 47.5% compared with 93% for Hodgkin's lymphoma.[7] In this study, disease was staged according to St. Judes classification and compliance/therapy was defined as adequate when enucleation was performed for stage 1 disease, chemoprevention was taken for at least four cycles for stage II disease and adjunctive chemotherapy was received for at least four cycles with radiotherapy in stage III retinoblastoma after enucleation/exenteration. The therapy was further considered effective only when four cycles of chemotherapy had been completed followed by an EUA after last the cycle and follow-up was available for at least 1 year after completion of therapy or till the occurrence of an event.[7] In a recent abstract presented at International Society of Pediatric Oncology , compliance was defined as per the ‘Pediatric Oncology in developing countries’ criteria.[53,54]
The delay in diagnosis needs be tackled at the level of primary pediatrician, where a red reflex test should be incorporated into each visit to the doctor. Compliance may be made better by improving coordination between the multiple specialties involved in the treatment of retinoblastoma, as frequent hospital visits, involvement of multiple specialties and longer duration of treatment are the probable reasons for poor compliance.
Footnotes
Source of Support: Nil