 PDF
PDF  Views
Views  Share
Share


Clinicopathoradiological findings in SEGA: A rare astroglial tumor
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2012; 33(03): 192-193
DOI: DOI: 10.4103/0971-5851.103158

Publication History
Article published online:
02 August 2021
© 2012. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Sir,
A 7½-year-old male presented with features of raised intracranial symptoms. He had a history of delayed milestones, mental retardation and seizures since 4 years. Examination revealed multiple facial angiofibromas [Figure 1]. Computed tomography (CT) imaging showed a large hyperintense lobular lesion, approximately 55 mm×50 mm in the right fronto-temporo-pareital region, causing compression over the frontal horn of the right lateral and third ventricles, with midline shift and subfalcine herniation to left, and hydrocephalous [Figure 2]. Following surgery, subsequent histopathological findings revealed closely packed polygonal to round cells with eosinophilic glassy hyaline cytoplasm and eccentrically positioned vesicular nuclei and prominent nucleoli in a fibrillary background [Figure 3]. Hence, the diagnosis of Subependymal giant cell astrocytoma (SEGA) was made. Following surgery, the patient was relieved of symptoms with no recurrence of tumor on follow-up.
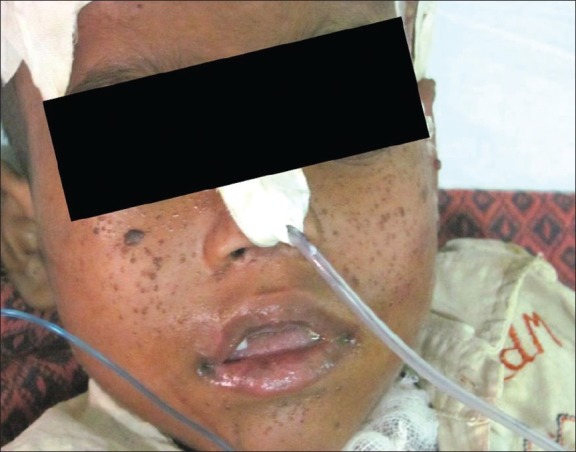
| Fig. 1 Clinical photograph demonstrating cutaneous adenoma sebaceum
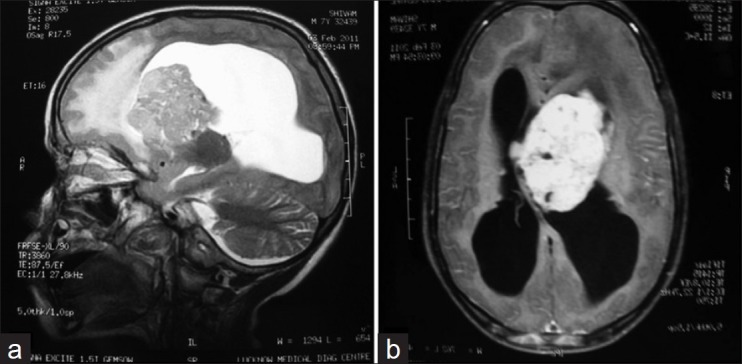
| Fig. 2 (a) T2 sagittal image: Large periventricular SOL is noted adjacent to the body and frontal horn of the left lateral ventricle. (b) T1 post-contrast axial image showing intraventricular extension of the same mass causing obstruction of the foramen of munro with non-communicating hydrocephalus

| Fig. 3 Microphotograph showing closely packed large polygonal to round cells with glassy hyaline eosinophilic cytoplasm and eccentric vesicular nucleus and prominent nucleoli in a fibrillary background (hematoxylin and eosin, ×100). Inset showing higher magnification (hematoxylin and eosin, ×400) of the same
Subependymal giant cell astrocytoma is a rare, slow-growing intraventricular tumor arising classically in 6-16% cases of tuberous sclerosis (TS).[1] TS is an autosomal dominant neurocutaneous phacomatosis with an incidence of 1 in 10,000. It comprises of hamartomatous tumors[2] and malformations affecting multiorgan systems, the central nervous system (CNS) being the most commonly involved. CNS lesions include cortical tubers, subependymal nodules, white matter heterotrophies and subependymal giant cell astrocytomas.[3] Other lesions include renal angiomyolipomas, cardiac rhabdomyomas and cystic lung disease, etc. Clinically, TS is characterized by Vogt's triad of mental retardation, seizures and adenoma sebaceum. SEGA are usually located in the lateral ventricles near the foramen of munro, causing obstructive hydrocephalus.[4,5] Radiological findings include sharply marginated and uniformly enhancing lesion on CT and magnetic resonance imaging, with hydrocephalus and size >1cm3. Grossly, it is a sharply delineated, spherical or multinodular mass of fleshy grey-pink tissue, anchored to the ventricular wall over a broad front. Tumor has a good prognosis, and recurrence is rare after surgical resection. Hence, the need for having a high index of suspicion and early identification of SEGA in patients of tuberous sclerosis can help prevent complications.
ACKNOWLEDGMENT
The author would like to thank Prof. Nuzhat Husain and Prof. B. K. Ojha for their helpful discussions. The author is also grateful to parents of the ailing child to provide consent for publication.
| Fig. 1 Clinical photograph demonstrating cutaneous adenoma sebaceum
| Fig. 2 (a) T2 sagittal image: Large periventricular SOL is noted adjacent to the body and frontal horn of the left lateral ventricle. (b) T1 post-contrast axial image showing intraventricular extension of the same mass causing obstruction of the foramen of munro with non-communicating hydrocephalus
| Fig. 3 Microphotograph showing closely packed large polygonal to round cells with glassy hyaline eosinophilic cytoplasm and eccentric vesicular nucleus and prominent nucleoli in a fibrillary background (hematoxylin and eosin, ×100). Inset showing higher magnification (hematoxylin and eosin, ×400) of the same