 PDF
PDF  Views
Views  Share
Share


Clinicohematological correlation and chromosomal breakage analysis in suspected Fanconi anemia patients of India
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2014; 35(01): 21-25
DOI: DOI: 10.4103/0971-5851.133706
Abstract
Introduction: The management of patients with aplastic anemia, to an extent, depends on the etiology i.e., inherited or acquired. The classical Chromosomal breakage study involves detection of chromosomal breakage or aberrations (breaks, gaps, rearrangements, radials, exchanges, endoreduplications) in peripheral blood cells after culture with a T-cell mitogen and a DNA clastogenic (cross-linking) agent, such as diepoxybutane (DEB) or mitomycin C (MMC). The testing needs to be performed in laboratory with appropriate expertise in Fanconi Anemia testing. The present study was undertaken to find out the frequency of inherited aplastic anemia in North India. Materials and Methods: This study was carried out at the Department of Molecular Biology and Transplant Immunology, Indraprastha Apollo hospital, New Delhi. The study includes retrospective analysis of 528 aplastic anemia patients whose samples were tested at our department for Chromosomal breakage study during the period 2007 to 2011. Respective age and sex matched healthy controls were also processed for chromosomal breakage study. Patient′s habitat, clinical symptoms, differential blood count and history of drug exposure were documented for all patients referred to us, whereever available. Relative risk was estimated by odds ratio (OR) with 95% confidence interval (CI) in matched cases and controls. Conclusion: A significant increase in chromosomal breakages was seen in 13.1% patients. The survival data documented for 100 patients suggested 60% mortality.
Keywords
Aplastic anemia - chromosomal anomalies - chromosomal breakage studies - Fanconis anemia - mitomycin-CPublication History
Article published online:
19 July 2021
© 2014. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Introduction:
The management of patients with aplastic anemia, to an extent, depends on the etiology i.e., inherited or acquired. The classical Chromosomal breakage study involves detection of chromosomal breakage or aberrations (breaks, gaps, rearrangements, radials, exchanges, endoreduplications) in peripheral blood cells after culture with a T-cell mitogen and a DNA clastogenic (cross-linking) agent, such as diepoxybutane (DEB) or mitomycin C (MMC). The testing needs to be performed in laboratory with appropriate expertise in Fanconi Anemia testing. The present study was undertaken to find out the frequency of inherited aplastic anemia in North India.
Materials and Methods:
This study was carried out at the Department of Molecular Biology and Transplant Immunology, Indraprastha Apollo hospital, New Delhi. The study includes retrospective analysis of 528 aplastic anemia patients whose samples were tested at our department for Chromosomal breakage study during the period 2007 to 2011. Respective age and sex matched healthy controls were also processed for chromosomal breakage study. Patient's habitat, clinical symptoms, differential blood count and history of drug exposure were documented for all patients referred to us, whereever available. Relative risk was estimated by odds ratio (OR) with 95% confidence interval (CI) in matched cases and controls.
Conclusion:
A significant increase in chromosomal breakages was seen in 13.1% patients. The survival data documented for 100 patients suggested 60% mortality.
INTRODUCTION
Bone marrow failure syndromes are disorders of hematopoietic stem cells that can lead to peripheral pancytopenia with marrow hypoplasia or aplastic anemia (AA). AA can be inherited or acquired.[1] Inherited disease includes Fanconi anemia (FA), dyskeratosis congenita, Shwachman-Diamond syndrome, Diamond-Blackfan anemia and amegakaryocytic thrombocytopenia.[2] FA is an autosomal recessive disease characterized by a variety of clinical symptoms and cellular features. Clinical manifestations are extremely pleomorphic and include bone marrow failure, congenital malformations, predisposition to various cancers, as well as chromosomal hypersensitivity to the clastogenic (chromosome-breaking) effect of cross-linking agents such as diepoxybutane (DEB) and mitomycin-C (MMC).[3] It is recommended that all patients exhibiting any congenital malformation known to be associated with FA or AA at any age, or any patient with myelodysplasia syndrome (MDS) with complex cytogenetic abnormalities, have a peripheral blood sample tested for cross-linker hypersensitivity. Due to the lack of concordance of FA phenotype among affected siblings, all full siblings of an FA patient should also be screened.[3]
Most of the Indian literature on this subject aims to detect a causative agents/presence of a genetic lesion, if any and development of molecular drug/s for the disease. The sample sizes in most of the studies, however been quite less.[4,5,6] Here, we describe a cohort of 528 patients of AA, who were subjected to FA tests on peripheral blood lymphocyte (PBL), with the aim of revealing FA cases and rule out this diagnosis in others. Clinical presentation and biological confirmation of FA patients identified are detailed and strategies for a comprehensive and precise diagnosis of FA in patients presenting with bone marrow failure (BMF) is discussed. We have also tried to throw some light on the demographic distribution of AA across India.
MATERIALS AND METHODS
Subjects
During the period from 2007 to 2011, 528 patients of AA were evaluated. A medical appointment with complete history and physical examination was performed by FA-experienced physicians and data was recorded. An informed consent was taken for each and every patient before he/she was subjected to a common questionnaire to draw the sketch of the clinical symptoms, etiological factor/s involved in and the pedigree of the patient. Patients included in the cohort were children or adults with bone marrow failure (at least one isolated or combined peripheral cytopenias and hypoplastic/aplastic bone marrow aspirates/biopsies). This included BMF patients (i) without any evidence of an associated underlying etiology, (ii) with only an isolated non-specific positive sign in history or physical examination (i.e., isolated short stature, or café-au-lait spots, or history of consanguinity) and (iii) patients with suspected genetic syndromes (based on clinical signs and/or family history), probably other than FA, who were tested to rule out the diagnosis of FA (iv) siblings of patients with a confirmed diagnosis of FA. (v) Known cases of FA. For all cases, cytopenia was defined as peripheral blood Hb <10 xss=removed>9/L and/or platelets <100 xss=removed>9/L.[7] Marrow hypoplasia/aplasia was defined based on standard histopathological diagnostic criteria. Patients further identified as having hypoplastic MDS were also analyzed.[8] The latest report of complete blood count and the details of whole blood and platelet transfusions of patients were taken to classify the severity of the disease. However, out of 528 patients, the latest blood counts of 256 patients were not available.
Chromosome breakage analysis by MMC method
A volume of 3-4 ml of peripheral blood sample collected in heparinized vacutainer (BD, vacutainers) were obtained from each patient. The cytogenetic diagnosis was conventionally made in PBL stimulated with phytohemagglutinin in 72 h cultures,[9] 0.5 ml of heparinized blood added to 5 ml of medium. Cultures were paired for MMC studies, with a replicate set of cultures to serve as untreated controls. MMC, at a final concentration in the medium of 100 ng/ml, was added to the treated cultures; untreated cultures were set and processed under the same conditions for routine karyotype analysis.
Control subjects were phenotypically normal individuals of both sexes ranging in age from 11 to 20 years, free of drugs, alcohol, or smoking habits who signed the informed consent to voluntarily participate in the study.[10] Slides were prepared and codified for the blind analysis of chromosome aberrations. Analysis was performed on 50 Giemsa-stained metaphases; each cell was scored for chromosome number and for the numbers and types of structural abnormalities [Figure 1].
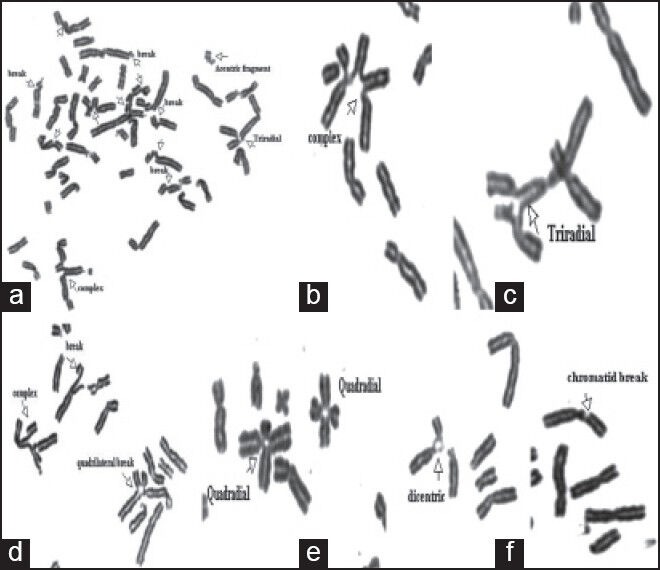
| Figure 1:Mitomycin-C-induced chromosomal aberrations in patients with Fanconi anemia (FA). (a-g) partial metaphases of FA patients: Chromosome break, acentric fragment, chromatid break, tri-radial, quadriradial
Achromatic areas, less than a chromatid in width, were excluded in the calculation of chromosome breakage frequencies, while exchange configurations, translocations, dicentric and ring chromosomes were scored as one chromosomal aberration. Radial configurations were scored separately. The figures are considered to be tri-radial when there are two breakages and tetraradial when there are three breakages.[11] The proportion of breaks and radial figures was expressed in percent, i.e., number of breaks or radial figures/number of mitotic figures ×100.[4] The definite diagnosis of FA was established when the PBL show a response to MMC, 3 times greater than a normal control.
RESULTS
Of 528 patients, the demographic data was available for only 310 (58.7%) patients. The patients belonged to 16 different states of northern India. Maximum patients of AA belonged to the states of Bihar, Delhi/NCR and Uttar Pradesh, accounting for 24%, 19.2% and 30.6% of the patient load respectively. The disease was most commonly seen in the patients that fall in the age group of 11-20 years.
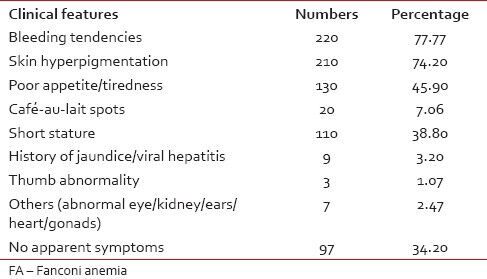
The age range of the patient was from the age of 1-45 year. However, 80.72% patients were <16 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4080657/#ref5" rid="ref5" class=" bibr popnode" role="button" aria-expanded="false" aria-haspopup="true" xss=removed>5] The clinical symptoms of the patient were recorded for 283 patients, including 60 (21.2%) patients with a positive chromosomal breakage analysis. The most common clinical presentation in these patients was bleeding tendencies (220, 77.7%) followed by skin hyperpigmentation (210, 74.2%) and poor appetite/tiredness (130, 45.9%) [Table 1]. No apparent symptoms were present in 97 (34.20%) patients including four patients (1.41%) who were MMC sensitive.
Table 1
Common clinical features in 283 suspected FA patients
|
The most common site of bleeding was through the nose (212, 96.3%). History of jaundice/viral hepatitis was a clinical presentation in 9 (3.2%) patients [Table 1]. Spontaneous breakage was observed in 3 (1.06%) patients and all were positive for chromosomal breakage analysis. AA was more common in patients from family with lower socio-economic status. The survival data documented for 100 patients suggest 60% mortality.
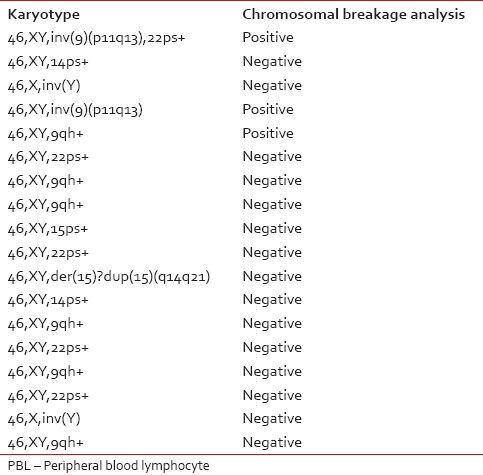
Of 528 patients, the chromosomal breakage study was possible only for 488 patients since 40 patients had culture failure due to one reason or the other, and a repeat sample could not be obtained. Out of these patients, 64 (13.1%) patients showed a significant increase in number of breaks in comparison to their control. The routine cytogenetic analysis was also performed on the peripheral blood samples of all 488 patients and revealed a normal karyotype in 470 (96.3%) patients and abnormal karyotype/polymorphic variants in 18 (3.7%) patients [Table 2]. Out of these 18 patients with abnormal karyotypes, 3 (16.7%) were found to be sensitive to MMC.
Table 2
Karyotype on routine cytogenetic analysis performed on PBL
Karyotype on routine cytogenetic analysis performed on PBL
|
DISCUSSION
FA, also known as Fanconi pancytopenia syndrome, is a rare and heterogeneous genetic disease affecting all ethnic groups and found in approximately 1 in 360,000 births.[11] Many genes can be responsible for this disease, which can be due to an autosomal recessive disease or an inherited X-linked disorder. However, all these genes have one thing in common that they do not allow the deoxyribonucleic acid (DNA) repair mechanisms work properly. These patients are at increased risk for both hematological and solid tumors (including leukemia, carcinomas and liver tumors).[11] Although, it is a gene disease instead of being a chromosomal disease, the diagnosis of FA is usually confirmed by a specific test known as chromosomal breakage study. It is a differential technique in which clastogenic substances such as DEB or MMC is used to promote DNA damage, breakage and rearrangement of chromosomes and cell death. Results are compared with those of a normal control group, especially matched for sex and age. The degree of sensitivity to DEB or MMC is correlated with neither the phenotype nor the severity of the disease. Furthermore, it is important to be aware that individuals that are heterozygous for the FA cannot be detected using this DEB/MMC test.[11]
The exact incidence of AA and its inherited form has not been published in India. Moreover, the sample size of these studies from India[6] has been very less and therefore, nothing could be predicted with confidence. The present study was conducted on 528 patients referred to our institution.
Of 528 patients, the cytogenetic classification for chromosomal breakage study was possible only for 488 patients, of them 64 (13.1%) patients showed a significant increase in the number of breaks in comparison to their control. This was in corroboration with other studies conducted in India.[5,6] In our study, we found that this disease is more common in northern states of India Viz. Bihar, Uttar Pradesh and Delhi. This could have been a biased observation as these places are in close proximity to Delhi where the study was being conducted. Therefore, a better approachability could have been the reason for the patients from this region coming for the test. The disease was common in the age group of 11-20-years-old patients whereas the study conducted by Varma et al.[6] showed a median age of 8 years for 54 cases of pediatric AA. This was also different from western literature where most of the patients are diagnosed between 3 and 7 years of age based on the presence of pancytopenia and only in 10%, the diagnosis is made after the age of 16.[10] According to the western literature, the male:female sex ratio is 1:1.[5] However, in the present study, the disease was found to be 3.4 times higher in male than female. The probable reason could be the male dominant social culture of the country. The complete blood count reports of 59% patients suggest severe condition of the disease. Most of these patients had bleeding tendencies and more specific features pertaining to FA were inexplicably low like thumb/radius abnormality or kidney abnormality, which are world-wide in the range of 35% in FA. Since these patients were also referred from other institutions; a regular follow-up and complete details in all the patients about their clinical features and their latest blood reports could not be obtained. However, patients appear to show a uniform trend in their clinic-hematological picture.
Limitations of the study include variable denominators for the study of these patients’ namely hematological parameters, clinical history for all patients and other epidemiological data availability for all patients.
Our results emphasize the importance of routinely conducting a diagnostic test in patients with AA. The role of FA laboratory in this regard is manifolds:
- An early clinical diagnosis of FA is both important mainly due to the complicated nature of the disease as well as its clinical manifestation being a part of other disorders. Many syndromes have been associated with this disease, and most of the clinical features overlap in one syndrome or the others. This extreme phenotypic diversity associated with FA makes the availability of a diagnostic laboratory test, especially valuable. Besides, early diagnosis of FA permits the exclusion of other diseases and precludes inappropriate management of hematologic disease (AA, MDS, acute myeloid leukemia) and permits appropriate consideration of stem cell transplant, androgens, hematopoietic growth factors or supportive care. In the present study, induced chromosomal breakage studies could diagnose FA in 64 patients. Although these patients were diagnosed after the onset of AA, MMC-stress test can be used to detect such patients even in the pre-anemic phase. This would help in avoiding drugs that are usually administered in acquired or ‘idiopathic’ AA. Further, screening parents of FA patients can help detect “silent” cases.
- Surgical intervention for orthopedic, renal or other anomalies is also optimized if the diagnosis of FA is known. For example, surgeries might be accelerated in order to be completed before the development of significant cytopenias or bleeding tendencies.
- Physicians can offer targeted cancer surveillance, counsel the patient well in advance, anticipate the problems that may happen as the disease progress and offer early, aggressive surgery for solid tumors.
- Genetic counseling is also imperative, because of the 25% risk of FA in each subsequent pregnancy.
- Information regarding MMC sensitivity is also extremely important in patients to be treated with bone marrow transplantation or chemotherapy. Since FA patients are hypersensitive to all DNA cross-linking agents, they require a modified pre-transplantation conditioning regimen, with a lower than the usual dose of cyclophosphamide or lower doses of chemotherapeutic agents.
Although the incidence of AA has increased in India over years, this disease has been overtly under diagnosed particularly its inherited form. The reasons for this include:
- Poor infrastructure and lack of expertise in FA testing in India.
- The socio-economic status and lack of affordability make it even more difficult to diagnose such patients.
- Male dominant society of the country.
- Lack of awareness and exposure about this rare disorder makes it even more difficult to estimate the real prevalence of this disease.
In the present study, we have tried to shed light on the health status of suspected FA patients, the pattern of disease, its occurrence, present status, distribution, etc., across the states in India with an emphasis on routinely conducting chromosomal breakage study in all patients with suspected FA.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Cirkoviæ S, Šæekiæ MG, Vujiæ D, Miæiæ D. Cytogenetic diepoxybutane sensitivity in Serbian children with Fanconi anemia. Arch Biol Sci Belgrad 2006;58:215-9.
- Pinto FO, Leblanc T, Chamousset D, Le Roux G, Brethon B, Cassinat B, et al. Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica 2009;94:487-95.
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009;668:4-10.
- Talwar R, Choudhry VP, Kucheria K. Differentiation of Fanconi anemia from idiopathic aplastic anemia by induced chromosomal breakage study using mitomycin-C (MMC). Indian Pediatr 2004;41:473-7.
- Jain D, Raina V, Fauzdar A, Mishra M, Tyagi N, Mahajan A, et al. Chromosomal breakage study in aplastic anemia patients in India. Asian J Med Sci 2010;2:227-32.
- Varma N, Varma S, Marwaha RK, Malhotra P, Bansal D, Malik K, et al. Multiple constitutional aetiological factors in bone marrow failure syndrome (BMFS) patients from north India. Indian J Med Res 2006;124:51-6.
- Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anemia: An International fanconi Anemia Registry study. Blood 1994;84:1650-5.
- Fohlmeister I, Fischer R, Mödder B, Rister M, Schaefer HE. Aplastic anaemia and the hypocellular myelodysplastic syndrome: Histomorphological, diagnostic, and prognostic features. J Clin Pathol 1985;38:1218-24.
- Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol 1993;21:731-3.
- Esmer C, Sánchez S, Ramos S, Molina B, Frias S, Carnevale A. DEB test for Fanconi anemia detection in patients with atypical phenotypes. Am J Med Genet A 2004;124A:35-9.
- Zen PR, Moraes FN, Rosa RF, Graziadio C, Paskulin GA. Clinical characteristics of patients with Fanconi anemia. São Paulo. Rev Paul Pediatr 2011;29:1-5.
| Figure 1:Mitomycin-C-induced chromosomal aberrations in patients with Fanconi anemia (FA). (a-g) partial metaphases of FA patients: Chromosome break, acentric fragment, chromatid break, tri-radial, quadriradial
References
- Cirkoviæ S, Šæekiæ MG, Vujiæ D, Miæiæ D. Cytogenetic diepoxybutane sensitivity in Serbian children with Fanconi anemia. Arch Biol Sci Belgrad 2006;58:215-9.
- Pinto FO, Leblanc T, Chamousset D, Le Roux G, Brethon B, Cassinat B, et al. Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica 2009;94:487-95.
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009;668:4-10.
- Talwar R, Choudhry VP, Kucheria K. Differentiation of Fanconi anemia from idiopathic aplastic anemia by induced chromosomal breakage study using mitomycin-C (MMC). Indian Pediatr 2004;41:473-7.
- Jain D, Raina V, Fauzdar A, Mishra M, Tyagi N, Mahajan A, et al. Chromosomal breakage study in aplastic anemia patients in India. Asian J Med Sci 2010;2:227-32.
- Varma N, Varma S, Marwaha RK, Malhotra P, Bansal D, Malik K, et al. Multiple constitutional aetiological factors in bone marrow failure syndrome (BMFS) patients from north India. Indian J Med Res 2006;124:51-6.
- Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anemia: An International fanconi Anemia Registry study. Blood 1994;84:1650-5.
- Fohlmeister I, Fischer R, Mödder B, Rister M, Schaefer HE. Aplastic anaemia and the hypocellular myelodysplastic syndrome: Histomorphological, diagnostic, and prognostic features. J Clin Pathol 1985;38:1218-24.
- Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol 1993;21:731-3.
- Esmer C, Sánchez S, Ramos S, Molina B, Frias S, Carnevale A. DEB test for Fanconi anemia detection in patients with atypical phenotypes. Am J Med Genet A 2004;124A:35-9.
- Zen PR, Moraes FN, Rosa RF, Graziadio C, Paskulin GA. Clinical characteristics of patients with Fanconi anemia. São Paulo. Rev Paul Pediatr 2011;29:1-5.