 PDF
PDF  Views
Views  Share
Share


Clinical implication of thiopurine methyltransferase polymorphism in children with acute lymphoblastic leukemia: A preliminary Egyptian study
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2015; 36(04): 265-270
DOI: DOI: 10.4103/0971-5851.171553
Abstract
Background: 6-mercaptopurine (6-MP) is an essential component of pediatric acute lymphoblastic leukemia (ALL) maintenance therapy. Individual variability in this drug-related toxicity could be attributed in part to genetic polymorphism thiopurine methyltransferase (TPMT). Aim: To investigate the frequency of common TPMT polymorphisms in a cohort of Egyptian children with ALL and the possible relation between these polymorphisms and 6-MP with short-term complications. Materials and Methods: This study included 25 children. Data related to 6-MP toxicity during the maintenance phase were collected from the patients′ files. DNA was isolated and genotyping for TPMT G460A, and A719G mutations were performed by polymerase chain reaction-restriction fragment length polymorphism. Results: Twenty (80%) of the included 25 patients had a polymorphic TPMT allele. TPMTFNx013A was the most frequent (14/25, 56%), 8 patients were homozygous and 6 were heterozygous. TPMTFNx013C mutant allele was found in 4 patients (16%) in the heterozygous state while 2 patients (8%) were found to be heterozygous for TPMTFNx013B mutant allele. TPMT mutant patients, especially homozygous, were at greater risk of 6-MP hematological toxicity without significant difference regarding hepatic toxicity. Conclusions: TPMT polymorphism was common among the studied group and was associated with increased risk of drug toxicity. A population-based multi-center study is required to confirm our results.
Keywords
6-mercaptopurine - acute lymphoblastic leukemia - pharmacogenetic - polymorphism - thiopurine methyltransferasePublication History
Article published online:
12 July 2021
© 2015. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Background:
6-mercaptopurine (6-MP) is an essential component of pediatric acute lymphoblastic leukemia (ALL) maintenance therapy. Individual variability in this drug-related toxicity could be attributed in part to genetic polymorphism thiopurine methyltransferase (TPMT).
Aim:
To investigate the frequency of common TPMT polymorphisms in a cohort of Egyptian children with ALL and the possible relation between these polymorphisms and 6-MP with short-term complications.
Materials and Methods:
This study included 25 children. Data related to 6-MP toxicity during the maintenance phase were collected from the patients’ files. DNA was isolated and genotyping for TPMT G460A, and A719G mutations were performed by polymerase chain reaction-restriction fragment length polymorphism.
Results:
Twenty (80%) of the included 25 patients had a polymorphic TPMT allele. TPMT*3A was the most frequent (14/25, 56%), 8 patients were homozygous and 6 were heterozygous. TPMT*3C mutant allele was found in 4 patients (16%) in the heterozygous state while 2 patients (8%) were found to be heterozygous for TPMT*3B mutant allele. TPMT mutant patients, especially homozygous, were at greater risk of 6-MP hematological toxicity without significant difference regarding hepatic toxicity.
Conclusions:
TPMT polymorphism was common among the studied group and was associated with increased risk of drug toxicity. A population-based multi-center study is required to confirm our results.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy.[1] Treatment success of this disease is attributed to adequate maintenance therapy necessary to prolong the obtained remission.[2] 6-mercaptopurine (6-MP) is a key component of pediatric ALL maintenance therapy.[3,4,5] It is a pro-drug that is activated by intracellular pathway[6] and then inactivated by either S-methylation via thiopurine methyltransferase (TPMT) or oxidation via xanthine oxidase.[7] Approximately 1 in 300 individuals have low TPMT activity, 6-11% have intermediate activity, and 89-94% have high activity.[8,9] Pharmacogenetic studies in childhood ALL have associated 6-MP efficacy and toxicity to single nucleotide polymorphism in genes coding for 6-MP metabolizing enzymes including TPMT.[10] Heterozygote individuals for TPMT mutations have intermediate activity while homozygote individuals have low activity.[11] More than 23 variants of the TPMT had been described.[12] The most common were TPMT*3C (A719G), TPMT*3B (G460A), TPMT*3A (G460A and A719G), and TPMT* (G238C), accounting for 89-95% of these inherited mutations.[12,13,14] There are substantial ethnic differences in the frequencies of low-activity variant alleles.[15]
We investigated the frequency of common TPMT polymorphisms in a cohort of ALL Egyptian children and the possible relation between these polymorphisms and 6-MP common short-term complications during the maintenance phase.
MATERIALS AND METHODS
This study included all newly diagnosed ALL children attending Hematology and Oncology Unit, Menoufia University Hospital, Egypt during the period from February 2010 to August 2012. All included children were native Egyptians, born and living in Menoufia governorate which is a rural area. The diagnosis of ALL was performed according to standard clinical, morphological, cytochemical, and immunophenotypic criteria. Patients were treated according to St. Jude ALL Total Therapy Study XV treatment protocol.[16] According to this protocol, the patients were categorized in risk groups depending on pretherapeutic factors and the response to the induction therapy. The study did not include patients with trisomy 21, patients on other therapy protocols, patients who failed to achieve remission after induction, those who relapsed or died before the first 20 weeks of maintenance phase, patients with impaired renal functions nor those with serological evidence of hepatitis C virus or hepatitis B viral infection at diagnosis or at any time during the follow-up phase. The rationale for the choice of 20 weeks completion of maintenance therapy was to obtain an adequate time for monitoring of toxicity.[17] The study included 25 ALL children (10 males and 15 females) with their age at diagnosis ranging from 1 to 14 years (6.4 ± 3.98, median 5). Twenty-three out of the 25 (93%) had common pre-B ALL and the other 2 (7%) had T type. According to the used protocol criteria for risk stratification, 12 patients (48%) were low risk while the remaining 13 (52%) were standard risk. The study was approved by the Research and Ethical Committee-Menoufia University. Informed consent from parents or guardians was collected for all children before blood sampling. In brief, St. Jude ALL Total Therapy Study XV treatment protocol consists of the following phases: A - Remission induction therapy (6 weeks). B - Consolidation treatment (8 weeks). C - Maintenance treatment (120 weeks for girls and 146 weeks for boys). Chemotherapy was given every week in full doses provided that the absolute neutrophil count was ≥300/mm3, white blood cells ≥1000/mm3, platelet count ≥50 × 109/L, with no evidence of mucositis, infection, or severe hepatotoxicity. Oral 6-MP in a dose of 75 mg/m2 was given daily in the evening on an empty stomach without milk. Doses of 6-MP were adjusted as per protocol. Patients missing ≥25% of therapy, as documented by missed treatment weeks, had 6-MP dosage reduced by 30% (from 75 to 50 mg/m2/day). The dose reduction was reviewed every 8 weeks.
This study is a descriptive in nature. Patients were followed till therapy completion or death or relapse. Blood sampling for TPMT polymorphism was done for patients when eligible for the toxicity analysis (after completion of 20 weeks of maintenance therapy). The blood samples were processed using serial numbers and kept until analysis. Relevant medical data of the maintenance phase were collected from the patients’ records. Data included 6-MP related adverse events (AEs), dose reductions, interruptions (number of weeks per cycle for 6-MP), and duration of 6-MP therapy at the time of sampling and at the time of appearance of adverse effects. The collected data were correlated with the specific TPMT genotypes after ending the time of follow-up. Hematologic and hepatic laboratory parameters related to 6-MP toxicity were monitored every 2 weeks during the maintenance phase and obtained from clinical charts and laboratory databases. AEs evaluation was assessed using the National Cancer Institute Common Terminology Criteria version 2.[18] For each withheld week of therapy due to toxicity caused by 6-MP, the main cause was documented (neutropenia or hepatotoxicity) and the week marked as a missed one for therapy.
We have investigated patients for TPMT variant alleles *3A, *3B, and *3C but not TPMT* based on its reported rarity.[19] Five milliliter of venous blood samples was withdrawn and transferred into ethylenediaminetetraacetic acid (EDTA) tubes. A unique accession number to each sample was assigned. For peripheral blood mononuclear cells separation, Lymphoflot solution was used (Bio test AG, Dreieich, Germany).[20] Genomic DNA was extracted using QIAamp DNA Blood Mini Kits (Qiagen, Hilden, Germany). DNA was diluted in 200 μL distilled water and stored at −20°C until analysis.
Thiopurine methyltransferase genotypes were determined by mutation-specific polymerase chain reaction (PCR). PCR amplification and enzymatic digestion of the products were performed by PCR-restriction fragment length polymorphism analysis to evaluate genetic polymorphism in TPMT (TPMT*3B [G460A], TPMT*3C [A719G], and TPMT*3A [G460A and A719G]) using method previously described.[21] For G460A polymorphism, genomic DNA (0.5–2 μg) was amplified with a forward primer 5′CAGGCTTTAGCATAATTTTCAATTCCTT-3′ and a reverse primer 5′-TGTTGGGATTACAGGTGTGAGCCAC-3′ (supplied by biosynthesis). While for A719G polymorphism, the forward primer was 5′CTT TGG GGA GCT GAA GGA CTA CTA C3′), and the reverse primer was 5′CAC TTT GTG ACC ATT CCG GTT TG 3′. PCR was performed with a Biometra thermo block (Biometra, Goettingen, Germany) using Perkin Elmer Thermal Cycler 2400 (Perkin Elmer, Waltham, Massachusetts, USA) under the standard conditions: An initial denaturation (3 min at 94°C), denaturation (1 min at 95°C), annealing (2 min at 55°C), extension (1 min at 72°C), and number of cycles: 40 with final extension (7 min at 72°C). The G460A polymorphism results in the creation of the MwoI recognition site while the A719G polymorphism results in the creation of the AccI recognition site. The products were run by 2% agarose gel electrophoresis for 30 min and stained with ethidium bromide, and the bands were visualized under ultraviolet light. For G460A polymorphism, wild-type GG showed two bands at 267 bp and 98 bp, heterozygous GA three bands of 365 bp, 267 bp, and 98 bp were seen and the homozygous AA had single band of 365 bp [Figure 1]. For A719G polymorphism, wild-type AA produced single fragments of 293 bp.
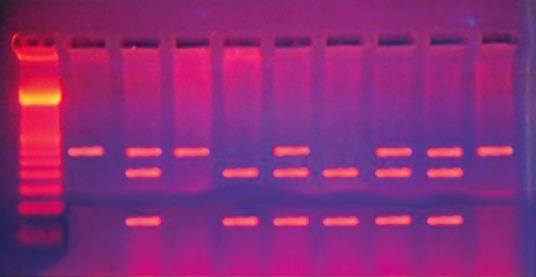
| Fig. 1 Electrophoreses pattern of 460G>A mutation
Heterozygous AG produced three fragments of 293, 207, and 86 bp, whereas, homozygous GG produced two fragments 207 and 86 bp [Figure 2]. Statistical analysis was performed using SPSS version 20 (SPSS Inc., Chicago, IL, USA). Data were expressed in mean ± standard deviation (SD). Differences between groups were analyzed using the Mann–Whitney U-test and frequencies by the Fischer exact test. P < 0.05 were considered statistically significant.
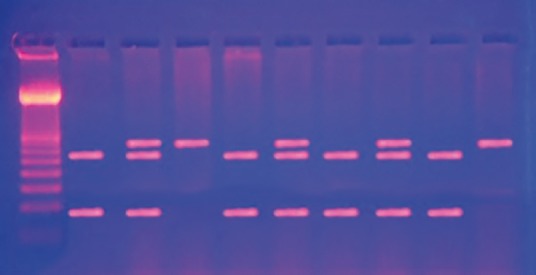
| Fig. 2 Electrophoreses pattern of the 719A>G mutation
RESULTS
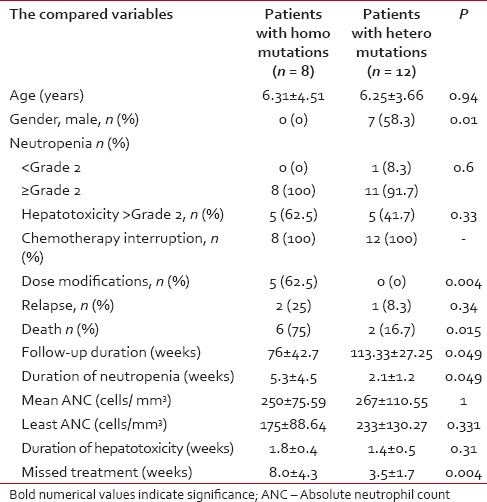
The observation period of the included patients ranged from 28 to 156 weeks, (mean ± SD of 95.2 ± 4.1 weeks, median 104). Eight out of 25 patients (32%) had completed their maintenance phase, 2 were males. During the study period, 4/25 ALL children (16%) had developed neutropenia < Grade 2 with or without fever while neutropenia of ≥Grade 2 (with or without fever) was traced in 21 patients (84%). 12/25 patients (48%) had developed ≥Grade 2 hepatotoxicity during their follow-up. Interruption of 6-MP treatment was reported in 23/25 patients (92%). The mean duration of 6-MP therapy interruption for the whole group was 4.92 ± 3.55 weeks (range of 0-16, median 4 weeks). According to the chemotherapy protocol, 6-MP dose modification was done for 5/25 (20%) patients while 20 (80%) did not need it. During the study duration, 3/25 (12%) patients relapsed (1 testicular, 1 central nervous system and 1 bone marrow). During the maintenance phase, 10 patients died – 3 due to relapse and 7 died due to neutropenic sepsis. TPMT genotyping revealed the presence of the mutant alleles in 20/25 ALL children while in 5 patients, none of the tested mutant alleles was found. TPMT*3A variant allele was found in 14 patients out of 20 (56%) with an allele frequency of 44%, 8 were homozygous TPMT*3A/TPMT*3A and 6 were heterozygous. For TPMT*3B mutant allele, 2 patients were found in a heterozygous state (allele frequency of 4%). On the other hand, TPMT*3C mutant allele was detected in 4 patients; all were in the heterozygous state (allele frequency of 8%). Patients carrying TPMT mutant alleles had significantly higher neutropenia ≥Grade 2 with infectious episodes, chemotherapy interruptions and mean duration of neutropenia compared to those without TPMT polymorphisms, without significant difference in the other parameters of toxicity including liver toxicity ≥Grade 2 [Table 1]. Patients with homozygous TPMT mutation had a significant higher percentage of dose modification, death, mean duration of neutropenia, and mean missed treatment weeks with significant lower duration of follow-up during the maintenance phase. The other parameters of toxicities did not differ between homozygous and heterozygous ALL children. All the carriers of homozygous mutation were females with a significant difference in gender distribution between homozygous and heterozygous [Table 2]. The three relapsed patients were carriers of the mutant TPMT*3A allele (2 homozygous and 1 heterozygous).
Table 1
Comparison of the toxicity between the mutant and nonmutant patients with leukemia during maintenance chemotherapy
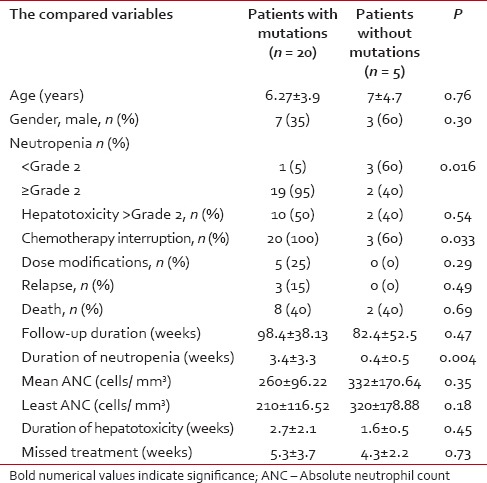
Table 2
Comparison of the toxicity between the homozygous mutant and the heterozygous mutant patients with leukemia during maintenance chemotherapy
DISCUSSION
Cross-ethnic differences in response to drugs have been related to the genotypic variation of the key enzymes and proteins that affect the safety and efficacy of a drug in the individual patient.[22] The prevalence of TPMT polymorphisms varies among different ethnic groups, ranging from 2% to 14%. TPMT*3A mutation accounts for more than 80% of the variant alleles in Caucasians, but for only approximately 17% of the variant alleles in African-Americans. On the other hand, TPMT*3C mutation accounts for only 5% of the variant alleles in Caucasians, 70% in African-Americans and 100% in the Japanese and Chinese.[23,24,25,26]
Among Arabs, Ayesh et al.[27] have reported that TPMT*3A was the only deficiency allele detected in the Palestinian ALL children with a frequency of 1.8% with an allelic frequency of 0.89%. Among 208 Tunisian subjects, TPMT*3C was the most common mutant allele accounting for 1.44%, followed by TPMT*3B in 0.24% where no TPMT*3A or TPMT* could be found.[28]
Data about TPMT polymorphism in Egypt are scarce. Furthermore, testing of TPMT polymorphism is not a routine work up in Egyptian cancer centers. Therefore, this study was conducted. The results of this work revealed that nonfunctional mutant TPMT alleles were common among the studied children (80%). TPMT*3A variant allele both in homozygous and heterozygous states was the most prevalent accounting for 70% (14/20) of the detected mutation followed by TPMT*3C (20%) then *3B (10%), a pattern that resembles Caucasians. These results are inconsistent with those of Hamdy et al.,[29] who had reported low frequency of TPMT variant alleles (6/200, 3%), with allele frequencies of 0.003 and 0.013 for TPMT*3A (14% of the mutant alleles) and TPMT*3C (86%), respectively, among healthy Egyptians. No TPMT*2 or *3B was detected in the included sample, a pattern which resembles Africans with respect to the TPMT*3C allele. In another study conducted on Egyptian ALL children, Aboul Naga et al.[30] had documented TPMT mutations in 23.1% (12/52) of the involved group, out of which both TPMT*2 and *3B were found in 9.6% in homozygous and heterozygous states; heterozygous TPMT*3A allele was found in 2 (3.8%) while no TPMT*3C was found. More recently, El-Kaffash et al.[31] reported TPMT*3A heterozygote state in 2.5% of the studied 40 standard risk B-cell ALL children. Among the involved 40 healthy children, TPMT*3A heterozygote state was found in 7.5% with 2.5% were heterozygotes for the TPMT*3C mutant allele.
It is notable that the frequency of TPMT mutant alleles found in our study (80%) is much higher than what has been reported in other similar previous studies performed in other ethnic groups,[23,24,25,26,32,33,34] among Arabs[27,28] and even among Egyptians.[29,30,31]
The high frequency of TPMT mutations and the difference in its pattern observed in this study could be related to the small sample size as well as to the difference of the localities from which the individuals were recruited which could be reflected on the ethnicity of our included children.
It is worth mentioning that Egypt is unique geographically, as it is located centrally between the three continents of Africa, Europe, and Asia, so its population is highly affected by the rapid pace of intercontinental transportation and large-scale immigration. Throughout history, the Greeks, Romans, Arabs, Turks, French, and British have all ruled Egypt and mixed with its people. Therefore, modern Egypt now is admixture of all these legacies.[29] In this study, all included ALL patients were nonrelated Egyptian children who were born and lived in Menoufia which is one of Nile Delta governorates of North Egypt. This may be a crucial difference since the previously mentioned Egyptian studies.[29,30,31] were done on urbanites who may have the have potential for gene admixture due to increased migration to the European countries.
Thiopurine methyltransferase polymorphism is an important determinant of 6-MP toxicity, even among patients who are heterozygous for this trait.[10] Hematological and hepatic toxicities are the most common adverse effects associated with cumulative toxic plasma concentrations of 6-MP metabolites[2,3] influenced by TPMT polymorphism.[7,11,35] This polymorphism greatly influences tolerance to 6-MP therapeutic regimen, even in heterozygous patients, for whom longer intervals of chemotherapy interruption were expected.[10,36] In this regard, this study revealed that the presence of TPMT mutation had a significant impact on hematological toxicity with the missed treatment weeks being more pronounced in homozygotes. Similar findings were detected in other studies.[10,11,17,30,33,36,37,38]
There is a debate about the etiological causes of hepatotoxicity among ALL patients under 6-MP therapy and its association with TPMT genotype. In this study, we did not detect any significant difference in hepatotoxicity regarding TPMT genotyping. This comes in accordance with Relling et al.,[10] who reported more frequent hepatotoxicity among those with wild genotype. Against this, significant association between hepatotoxicity and TPMT genotyping was documented in previous studies.[30,33]
Despite the previous documentation of the significant implication of TPMT genotype and 6-MP related toxicity, there are some studies that did not detect this significant association.[27,32,39,40]
If treated with standard doses of thiopurines, TPMT deficient patients accumulate excessive thioguanine nucleotides in hematopoietic tissues, leading to severe hematological toxicity that could be fatal, with those who are homozygous will be at greater risk for a poor outcome from ALL therapy.[10] In agreement with what has been mentioned, although the survival analysis was not included, this study documented a significant higher death rate among homozygous mutant patients.
6-mercaptopurine dose intensity is clarified as an important determinate of event-free survival in childhood leukemia.[36] Higher 6-TGNs levels, due to TPMT mutant genotype, are associated with increased myelosuppression but the patients may get the benefit of decreasing the associated relapse due to increased dose intensity of more intracellular 6-TGNs inside the tumor cells.[41] Preliminary data indicate that minimal residual disease positivity on day 78 of treatment was significantly lower in heterozygotes than in patients with homozygous wild-type alleles.[42] However, in our study, TPMT polymorphism had no significant impact on relapse rate during the maintenance phase. The same was reported in previous studies.[37,40]
CONCLUSION
Despite the small numbers and a single locality, this study revealed a noteworthy result of high frequency of TPMT mutant alleles among the studied ALL children with a unique pattern. TPMT polymorphism increased the risk for 6-MP related hematological toxicity and its fatal outcome that was more prominent in the homozygous form. This emphasizes the importance of common TPMT variants determination in ALL children prior to therapy which could help tailoring 6-MP doses and reducing the associated toxicity. Nevertheless, a large population-based study in multiple Egyptian centers is required to confirm our results.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
| Fig. 1 Electrophoreses pattern of 460G>A mutation
| Fig. 2 Electrophoreses pattern of the 719A>G mutation