 PDF
PDF  Views
Views  Share
Share


Choroid Plexus Carcinoma Enmasked with Soft Signs and Symptoms
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2019; 40(04): 584-587
DOI: DOI: 10.4103/ijmpo.ijmpo_83_18
Abstract
Choroid plexus carcinomas (CPCs) are rare brain tumors, with preponderance in infants. They are highly invasive tumors with a dismal prognosis. The 5-year survival rates for CPC vary between 10% and 50%. They commonly arise from the lateral ventricles. CPCs in infants present with the subtle features of raised intracranial tension (ICT). Surgical resection is the mainstay of management with adjuvant chemotherapy and radiotherapy. We report a case of 11-month-old child with CPC, presented with subtle nonspecific signs and symptoms of raised ICT, treated by surgical resection and chemotherapy.
Publication History
Received: 11 April 2018
Accepted: 12 September 2018
Article published online:
03 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Choroid plexus carcinomas (CPCs) are rare brain tumors, with preponderance in infants. They are highly invasive tumors with a dismal prognosis. The 5-year survival rates for CPC vary between 10% and 50%. They commonly arise from the lateral ventricles. CPCs in infants present with the subtle features of raised intracranial tension (ICT). Surgical resection is the mainstay of management with adjuvant chemotherapy and radiotherapy. We report a case of 11-month-old child with CPC, presented with subtle nonspecific signs and symptoms of raised ICT, treated by surgical resection and chemotherapy.
Introduction
Choroid plexus carcinomas (CPCs) are rare intracranial neoplasms arising from neuroepithelial lining of the choroid plexus. They account for only 0.3% of all the intracranial tumors.[1] Choroid plexus tumor (CPT) was first described by Guerard in 1833. In 1906, Bielschowsky Unger reported first surgical resection. Accounting for 20%–30% of CPT, choroid plexus carcinomas are malignant counterpart of choroid plexus papilloma (CPP). About 70% of CPC are commonly seen in children, which occur within the first 2 years of life. In children, they are mostly supratentorial in origin. Around 50% of the CPC arise from lateral ventricles.[2] Signs and symptoms of raised intracranial tension (ICT) are the common clinical manifestations in children.[1] [2] [3] CPTs are classified on the basis of their histological criteria. Surgical resection is the mainstay of management and for prognostication. Chemotherapy and radiotherapy are adjuvant treatment. Recurrence rate of the tumor accounts for 13% and 5-year survival rate is only 40% with the multimodal therapy. Survivors of CPCs have some neurological compromise depending on the extent of invasion.[4] [5] Presentation with atypical symptoms and signs is not frequently encountered for this entity. Here, we report a 11-month-old male child with CPC presenting with atypical signs and symptoms.
Case Report
A firstborn, 11-month-old male child, born to nonconsanguineously married couple presented to our emergency unit with the history of loose stools for 7 days, fever, refusal of feeds, irritability, and excessive cry for 2 days. On probing, the child had history of irritability, excessive cry, and lethargy for last 6 weeks for which child had multiple hospital visits and treated symptomatically. The child had normal development. Birth history and postnatal course were uneventful. At admission, child was sick and irritable and Glasgow Coma Scale was 10/15 with stable vitals. General physical examination showed Grade 2 pallor, anterior fontanelle was bulged and tense [Figure 1]. There were no neurocutaneous markers. Head circumference and Anthropometric measurements were normal. On neurological examination, pupils were equal and reactive to light, extraocular movements and fundoscopy were normal. No motor or sensory deficits were noted. Other systemic examination was unremarkable. Provisional diagnosis of acute encephalitis syndrome with raised ICP was made and treated as per protocol. Investigations showed microcytic hypochromic anemia with neutrophilic leukocytosis and thrombocytosis. C-reactive protein was elevated. Guarded lumbar puncture done revealed hypoglycorrhachia, elevated proteins and two lymphocytes with no malignant cells. Computed tomography (CT) brain was suggestive of intracranial space-occupying lesion arising from the periventricular region [Figure 2]. Contrast-enhanced magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) [Figure 3] and [4] showed large lobulated avidly enhancing intraventricular mass arising from the right ventricle (7.3 cm × 7.9 cm × 8.1 cm) with multifocal internal necrotic component, calcific foci, and intrinsic flow voids. There was extensive white matter edema with mass effects in the form of effacement of cortical sulci, right uncal herniation, and midbrain/third ventricular compression. Both optic nerves were torturous with indenting optic disc. MRS revealed markedly elevated choline/creatinine ratio with NAA and elevated lipid/lactate peak at 1.3 ppm, all features consistent of diagnosis of CPC.
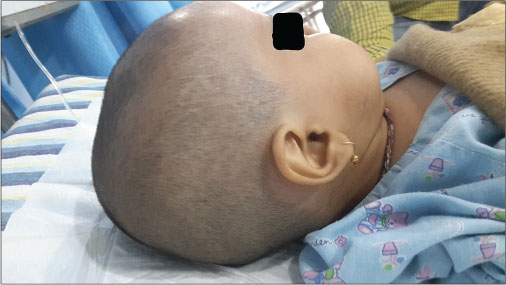
| Fig. 1 Bulging anterior fontanel
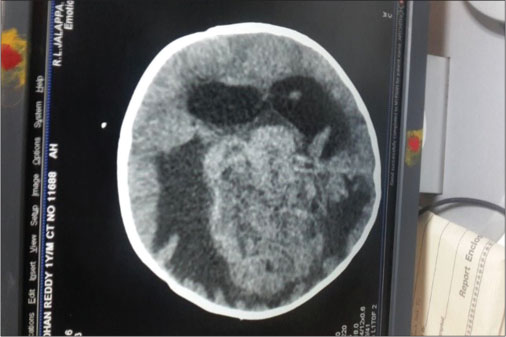
| Fig. 2 Intracranial space-occupying lesion from periventricular region
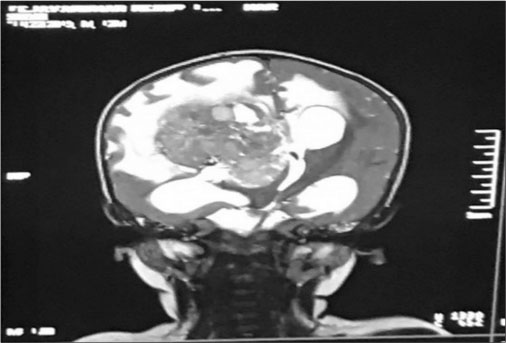
| Fig. 3 T2 Coronal sequence post contrast shows large lobulated avidly enhanching intraventricular mass arising from right ventricle with multifocal internal cystic/necrotic component, calcific foci and intrinsic flow voids
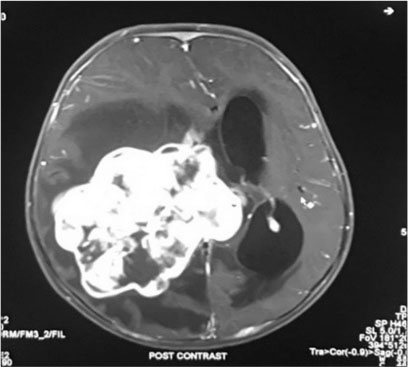
| Fig. 4 T1 fat suppressed post contrat axial study shows intraventricular mass from right ventricle with midline shift to the left and mild hydrocephalus
The child was referred to the neurosurgical center, where right temporoparietal craniotomy and decompression of the lesion were done. Grossly, lesion was grayish firm and easily suckable and mildly vascular in the right temporoparietal region. Histopathological examination (HPE) was suggestive of WHO Grade III CPC [Figure 5] and [6]. The child was transferred to the oncological center where adjuvant chemotherapy with vincristine, cyclophosphamide, and alternating with carboplatin and etoposide once in 3 weeks is being continued. Radiotherapy was deferred in view of his age.
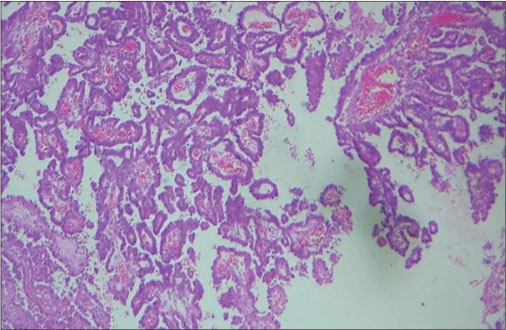
| Fig. 5 Papillae lined by neoplastic cells (H and E, ×100)
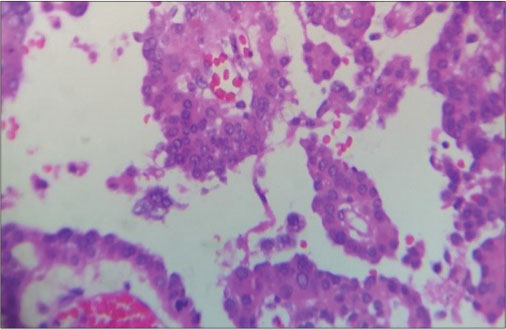
| Fig. 6 The nuclei show pleomorphism with mitoses
Follow-up MRI, 9 months following surgery, showed no obvious enhancing lesion. Presently, the child is visually handicapped secondary to optic atrophy and has been attaining motor and language milestones appropriate for age with no signs of recurrence.
Discussion
CPTs are rare intraventricular papillary neoplasms derived from the neuroectodermal epithelium, which account for approximately 2%–4% of intracranial tumors in children. CPP are benign and more common than CPCs in a ratio of 5:1. CPCs account for 0.6% of all brain tumors. Approximately 80% of CPCs arise in children, and 20% are seen in adults. The average annual incidence is 0.3 per 1,000,000 population. They are more common in young children, with a median age diagnosis of 1.5 years in case of supratentorial tumors. In comparison in adults CPCs are diagnosed as infratentorial fourth ventricle or cerebellopontine tumors.[1] [2] [3]
Etiology of CPC’s is unknown. They are sporadic in origin. Majority of CPC’s harbor TP53 mutations, and Merino et al. observed that tumors with increased mutant copies are the most likely to be aggressive with poorer survival.[6] Notch 3 signaling, the transcription factor TWIST1, platelet-derived growth factor receptor, and the tumor necrosis factor related apoptosis-inducing ligand pathway have also been implicated in papilloma tumorigenesis.[6] [7] CPCs have been associated with Li–Fraumeni syndrome, with mutation of p53 tumor-suppressor gene, Aicardi syndrome and possible association with Simian Virus 40 is observed since DNA sequences of virus have been identified in the tumor’s genome.[7]
At diagnosis, CPCs vary significantly in size. CPCs are softer and more friable than papilloma’s on gross, but the grade of neoplasm cannot be differentiated. Although carcinomas rarely metastasize, they disseminate throughout the cerebrospinal fluid (CSF) pathways. Definitive diagnosis is usually based on histopathology. There are three major features associated with malignancy.[8] First, cytologic criteria, such as nuclear atypia, increased nuclear: cytoplasmic ratio and prominent mitotic figures. Second, there is loss of normal papillary architecture. The third is the presence of brain invasion by the tumor, which usually involves extension through ependymal lining. As per recent WHO classification of tumors of central nervous system, CPTs are considered as distinct variety of neuroepithelial tumors and are subclassified as CPP (Group I), atypical CPP (Group II), or CPC (Group III).[5] [9]
On immunohistochemistry positive reactivity for cytokeratin, vimentin, S-100 protein, transthyretin, and carboxy anhydrase but not for epithelial membrane antigen, typifies CPCs.[7] Recently, use of transthyretin as an immunohistochemical marker aids in the distinction of primary CPT from other tumors that may invade the choroid plexus. The absence of transthyretin in tumor cells is associated with poor prognosis. Other poor prognosticators include decreased immunoreactivity with S100 protein, brain invasion by neoplastic cells, absence of stromal edema, and necrotic areas. Elevated mitotic activity (more than 2 mitoses per 20 HPF) was the sole feature independently associated with higher likelihood of recurrence.[2] [6]
In children, lateral ventricles are the common sites of CPCs accounting for 80%. Children with CPCs have varied clinical symptomatology and present with signs and symptoms of raised ICP, such as vomiting, headaches, and unsteadiness. As the median age of onset is 9 months, macrocephaly is often present, especially in congenital tumors as reported by Humphreys et al.[1] Hydrocephalus is believed to be secondary to the overproduction of CSF. There may be secondary hemorrhage within the CSF and increased ventricular dilatation due to subarachnoid reabsorption block.[2] Children usually have abrupt onset of symptoms before diagnosis, mostly ranging from 1 week to 2 months due to their chronicity. Carcinomas tend to be more locally invasive and frequently have focal neurologic deficits without hydrocephalus early in the illness. Metastatic spread is more common in CPCs and the dissemination is found on staging studies.[2] [3]
Neuroradiologically CPTs are indistinguishable. CT and MRI demonstrate the invasion of CPCs into the surrounding brain with peritumoral vasogenic edema. CPCs on contrast-enhanced CT are noted as isodense to hyperdense heterogeneous mass with intense homogeneous enhancement of solid portions. MRI shows hypointense on T1-weighted images. On T2-weighted images, the solid portion of the tumor may have associated cystic or necrotic regions. Arteriography may be valuable as part of an intravascular embolization procedure to reduce blood flow to the tumor, before surgical excision.[10]
There is lack of definitive guidelines on the management in CPCs. Literature shows that gross total resection (GTR) has favorable impact on survival, but it is achieved in < 50% of cases. Combined with adjunctive therapy, survival ranges from 67% to 91%. Surgical challenges faced include increased tumor vascularity, lack of well-developed plane between the brain and tumor, and excessive friability of the tumor.[4] Wrede et al. conducted a study on patients with CPC, comparing those who underwent complete resection versus subtotal resection. 2-year survival rate was 72.9% for patients with complete resection and 41.9% with partial resection, which demanded a second surgery.[11] Most chemotherapy regimens rely on cyclophosphamide, etoposide, vincristine, and platinum. Wolff and colleagues had disappointing results on treating with chemotherapy alone. Adjunctive chemotherapy with or without radiotherapy after an initial surgical procedure has found to be effective.
Prognosis of children with CPCs is poorer than those with papillomas. When treated with GTR over two-thirds of children had disease-free survival at 5 years as observed by Sun et al.[5] and recurrence was noted in 13%. The need for adjuvant therapy has been documented with partial resection. Children may have significant residual neurologic squeal such as hemiparesis, seizure disorders, and cognitive dysfunction, especially with tumors that have invaded eloquent areas of the brain.
Conclusion
CPCs are rare aggressive tumors predominantly seen in infancy. Atypical symptoms and subtle signs of raised ICT in children usually lead to delay in the diagnosis, with presentation in late stages of disease. Due to indistinguishable features from CPP on neuroimaging, histopathology upfronts the definitive diagnosis. The extent of surgical resection of the tumor forms the best prognosticating marker. Adjuvant chemotherapy and radiotherapy increase the 5-year survival rates up to 60%–90%. There is a need to keep a high suspicion of intracranial tumors in infants presenting with subtle symptoms and signs of raised intracranial pressure and a continuous follow-up for early detection of recurrence and appropriate management of neurological sequelae.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given his consent for images and other clinical information to be reported in the journal. The Guardian understands that name and initials will not be published and due efforts will be made to conceal patient identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- 1 Levy M, Goldfarb A, Hyder D. Choroid plexus tumors in children: Significance of stromal invasion. Neurosurgery 2001; 48: 303-9
- 2 Filippidis AS, Tsonidis CA. Intra ventricular brain tumors in children. Pediatr Neurosurg 1989; 20: 230-3
- 3 Geerts Y, Gabreels F, Lippens R, Merx H, Wesseling P. Choroid plexus carcinoma: A report of two cases and review of the literature. Neuropediatrics 1996; 27: 143-8
- 4 McEvoy AW, Harding BN, Phipps KP, Ellison DW, Elsmore AJ, Thomson D. et al. Management of choroid plexus tumours in children: 20 years’ experience at a single neurosurgical centre. Pediatr Neurosurgery 2000; 32: 192-9
- 5 Sun MZ, Ivan ME, Clark AJ, Oh MC, Delance AR, Oh T. et al. Gross total resection improves overall survival in children with choroid plexus carcinoma. J Neurooncol 2014; 116: 179-85
- 6 Merino DM, Shlien A, Villani A, Pienkowska M, Mack S, Ramaswamy V. et al. Molecular characterization of choroid plexus tumors reveals novel clinically relevant subgroups. Clin Cancer Res 2015; 21: 184-92
- 7 Safaee M, Oh MC, Bloch O, Sun MZ, Kaur G, Auguste KI. et al. Choroid plexus papillomas: Advances in molecular biology and understanding of tumorigenesis. Neuro Oncol 2013; 15: 255-67
- 8 Ogiwara H, Dipatri Jr. AJ, Alden TD, Bowman RM, Tomita T. Choroid plexus tumors in pediatric patients. Br J Neurosurg 2012; 26: 32-7
- 9 Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. et al. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016; 131: 803-20
- 10 Fitzpatrick LK, Aronson LJ, Cohen KJ. Is there a requirement for adjuvant therapy for choroid plexus carcinoma that has been completely resected?. J Neurooncol 2002; 57: 123-6
- 11 Wrede B, Liu P, Ater J, Wolff JE. Second surgery and the prognosis of choroid plexus carcinoma-results of a meta-analysis of individual cases. Anticancer Res 2005; 25: 4429-33
Address for correspondence
Dr. KNV PrasadDepartment of Paediatrics, Sri Devraj Urs Medical CollegeKolar - 563 101, KarnatakaIndiaEmail: drknvp@gmail.comPublication History
Received: 11 April 2018
Accepted: 12 September 2018
Article published online:
03 June 2021© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig. 1 Bulging anterior fontanel
| Fig. 2 Intracranial space-occupying lesion from periventricular region
| Fig. 3 T2 Coronal sequence post contrast shows large lobulated avidly enhanching intraventricular mass arising from right ventricle with multifocal internal cystic/necrotic component, calcific foci and intrinsic flow voids
| Fig. 4 T1 fat suppressed post contrat axial study shows intraventricular mass from right ventricle with midline shift to the left and mild hydrocephalus
| Fig. 5 Papillae lined by neoplastic cells (H and E, ×100)
| Fig. 6 The nuclei show pleomorphism with mitoses
References
- 1 Levy M, Goldfarb A, Hyder D. Choroid plexus tumors in children: Significance of stromal invasion. Neurosurgery 2001; 48: 303-9
- 2 Filippidis AS, Tsonidis CA. Intra ventricular brain tumors in children. Pediatr Neurosurg 1989; 20: 230-3
- 3 Geerts Y, Gabreels F, Lippens R, Merx H, Wesseling P. Choroid plexus carcinoma: A report of two cases and review of the literature. Neuropediatrics 1996; 27: 143-8
- 4 McEvoy AW, Harding BN, Phipps KP, Ellison DW, Elsmore AJ, Thomson D. et al. Management of choroid plexus tumours in children: 20 years’ experience at a single neurosurgical centre. Pediatr Neurosurgery 2000; 32: 192-9
- 5 Sun MZ, Ivan ME, Clark AJ, Oh MC, Delance AR, Oh T. et al. Gross total resection improves overall survival in children with choroid plexus carcinoma. J Neurooncol 2014; 116: 179-85
- 6 Merino DM, Shlien A, Villani A, Pienkowska M, Mack S, Ramaswamy V. et al. Molecular characterization of choroid plexus tumors reveals novel clinically relevant subgroups. Clin Cancer Res 2015; 21: 184-92
- 7 Safaee M, Oh MC, Bloch O, Sun MZ, Kaur G, Auguste KI. et al. Choroid plexus papillomas: Advances in molecular biology and understanding of tumorigenesis. Neuro Oncol 2013; 15: 255-67
- 8 Ogiwara H, Dipatri Jr. AJ, Alden TD, Bowman RM, Tomita T. Choroid plexus tumors in pediatric patients. Br J Neurosurg 2012; 26: 32-7
- 9 Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. et al. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016; 131: 803-20
- 10 Fitzpatrick LK, Aronson LJ, Cohen KJ. Is there a requirement for adjuvant therapy for choroid plexus carcinoma that has been completely resected?. J Neurooncol 2002; 57: 123-6
- 11 Wrede B, Liu P, Ater J, Wolff JE. Second surgery and the prognosis of choroid plexus carcinoma-results of a meta-analysis of individual cases. Anticancer Res 2005; 25: 4429-33