 PDF
PDF  Views
Views  Share
Share


CD4+/CD8- T cell Large Granular Lymphocytic Leukemia: A rare Entity
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2019; 40(03): 453-455
DOI: DOI: 10.4103/ijmpo.ijmpo_19_18
Sir,
Large granular lymphocytic leukemia (LGL) is a well-recognized disorder of mature T-cells or NK cells. T-cell LGL leukemia (T-LGL) is characteristically a disorder of mature CD3+/CD8+ cytotoxic T-cells. Rare variants include CD 3+/CD4+/CD8-cases. To the best of our knowledge, 11 such cases (4 cases by Lima et al.[1] in 2003, 4 cases by Olteanu et al. in 2010,[2] 2 cases by Mutreja et al.[3] in 2014, and 1 case by Rabade et al.[4] in 2014) of T-LGL showing CD3+/CD4+/CD8-immunophenotype has been published in literature so far. There is a paucity of literature explaining the monoclonal expansion of CD3+/CD4+ T-LGL.[1] Unlike CD8+ T-LGL, CD4+ T-LGL does not show cytopenia, autoimmune phenotypes,[1],[5] or splenomegaly. However, CD4+ T-LGL is frequently associated with nonhematological malignancies.[1] Here, we report a case presenting with CD3+/CD4+/CD8-immunophenotype. Such immunophenotypic variant form of T-LGL cases should have a close clinical follow-up as they are prone to develop either simultaneously or months and years after, secondary hematological or nonhematological malignancies.[1]
We received a peripheral blood sample for immunophenotyping from a 51-year-old female with a 4-month history of persistent lymphocytosis. Clinical examination revealed a single cervical lymphadenopathy with no hepatosplenomegaly. The complete blood count showed mild anemia (hemoglobin - 11.9 g/dL), a normal platelet count (platelets 150,000/μL), and absolute lymphocytosis (total leukocyte count 13.0 × 103/μL with 79.5% lymphocytes). Peripheral smear examination revealed a large number of large granular lymphocytes. Cytogenetic analysis was not performed.
Immunophenotyping of peripheral blood was carried out using the lyse wash method and a four-color flow cytometry panel [Table 1]. The antibody clones used are shown in [Table 2]. The sample was run on a BDFACS Calibur instrument (Becton/Dickinson Biosciences), and the immunophenotyping data were analyzed with BD Cell Quest software. The percentage of positive cells above a threshold set against a processed isotype control tube was used to express the florescence measurement. Flow cytometry analysis of a heparin peripheral blood sample showed a large lymphoid cell cluster (62% of total cells) with bright CD45 positivity. The cells showed positivity for CD3 (96%), CD4 (94%), CD5 (96%), CD2 (97%), CD16 (41%), CD56 (90%), and-CD57 (91%), indicating a T-cell origin [Figure 1]. There was an aberrant loss of CD7 expression, and CD8 expression was negative [Figure 1]. Other B lymphoid cells markers were negative including CD10, CD19, CD23, CD20, CD38, and surface kappa/lambda light chain expression. In accordance with morphology [Figure 2] and immunophenotypic findings, a laboratory diagnosis of CD3+/CD4+/CD8-T-LGL was established. The patient was monitored, and no chemotherapy was administered. On follow-up at 6 months, the patient was asymptomatic with persistent cervical lymphadenopathy.
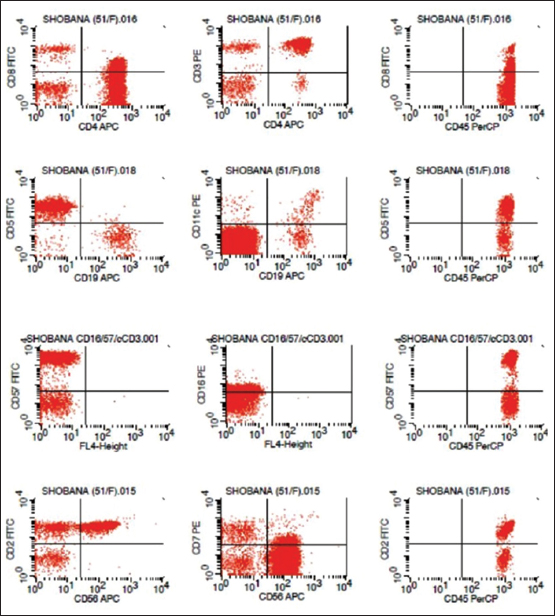
| Figure 1 The cells showed positivity for CD3, CD4, CD5, CD2, CD16, CD56, and - CD57, indicating a T-cell origin. There was aberrant loss of CD7 expression, and CD8 expression was negative

| Figure 2 Giemsa stained peripheral blood film (×100) showing large granular lymphocytes
|
Fluorochromes |
FITC |
PE |
PerCP |
APC |
|---|---|---|---|---|
|
Tube1 |
lgG1 |
lgG1 |
CD45 |
lgG1 |
|
Tube2 |
CD2 |
CD7 |
CD45 |
CD56 |
|
Tube3 |
CD8 |
CD3 |
CD45 |
CD4 |
|
Tube4 |
CD10 |
CD20 |
CD45 |
Blank |
|
Tubes |
CD5 |
CD11c |
CD45 |
CD19 |
|
Tube6 |
FMC7 |
CD23 |
CD45 |
CD20 |
|
Tube7 |
sKappa |
slambda |
CD45 |
CD19 |
|
Tube8 |
CD57 |
CD16 |
CD45 |
Blank |
Sir,
Large granular lymphocytic leukemia (LGL) is a well-recognized disorder of mature T-cells or NK cells. T-cell LGL leukemia (T-LGL) is characteristically a disorder of mature CD3+/CD8+ cytotoxic T-cells. Rare variants include CD 3+/CD4+/CD8-cases. To the best of our knowledge, 11 such cases (4 cases by Lima et al.[1] in 2003, 4 cases by Olteanu et al. in 2010,[2] 2 cases by Mutreja et al.[3] in 2014, and 1 case by Rabade et al.[4] in 2014) of T-LGL showing CD3+/CD4+/CD8-immunophenotype has been published in literature so far. There is a paucity of literature explaining the monoclonal expansion of CD3+/CD4+ T-LGL.[1] Unlike CD8+ T-LGL, CD4+ T-LGL does not show cytopenia, autoimmune phenotypes,[1],[5] or splenomegaly. However, CD4+ T-LGL is frequently associated with nonhematological malignancies.[1] Here, we report a case presenting with CD3+/CD4+/CD8-immunophenotype. Such immunophenotypic variant form of T-LGL cases should have a close clinical follow-up as they are prone to develop either simultaneously or months and years after, secondary hematological or nonhematological malignancies.[1]
We received a peripheral blood sample for immunophenotyping from a 51-year-old female with a 4-month history of persistent lymphocytosis. Clinical examination revealed a single cervical lymphadenopathy with no hepatosplenomegaly. The complete blood count showed mild anemia (hemoglobin - 11.9 g/dL), a normal platelet count (platelets 150,000/μL), and absolute lymphocytosis (total leukocyte count 13.0 × 103/μL with 79.5% lymphocytes). Peripheral smear examination revealed a large number of large granular lymphocytes. Cytogenetic analysis was not performed.
Immunophenotyping of peripheral blood was carried out using the lyse wash method and a four-color flow cytometry panel [Table 1]. The antibody clones used are shown in [Table 2]. The sample was run on a BDFACS Calibur instrument (Becton/Dickinson Biosciences), and the immunophenotyping data were analyzed with BD Cell Quest software. The percentage of positive cells above a threshold set against a processed isotype control tube was used to express the florescence measurement. Flow cytometry analysis of a heparin peripheral blood sample showed a large lymphoid cell cluster (62% of total cells) with bright CD45 positivity. The cells showed positivity for CD3 (96%), CD4 (94%), CD5 (96%), CD2 (97%), CD16 (41%), CD56 (90%), and-CD57 (91%), indicating a T-cell origin [Figure 1]. There was an aberrant loss of CD7 expression, and CD8 expression was negative [Figure 1]. Other B lymphoid cells markers were negative including CD10, CD19, CD23, CD20, CD38, and surface kappa/lambda light chain expression. In accordance with morphology [Figure 2] and immunophenotypic findings, a laboratory diagnosis of CD3+/CD4+/CD8-T-LGL was established. The patient was monitored, and no chemotherapy was administered. On follow-up at 6 months, the patient was asymptomatic with persistent cervical lymphadenopathy.
| Figure 1 The cells showed positivity for CD3, CD4, CD5, CD2, CD16, CD56, and - CD57, indicating a T-cell origin. There was aberrant loss of CD7 expression, and CD8 expression was negative
| Figure 2 Giemsa stained peripheral blood film (×100) showing large granular lymphocytes
Fluorochromes |
FITC |
PE |
PerCP |
APC |
|---|---|---|---|---|
|
Tube1 |
lgG1 |
lgG1 |
CD45 |
lgG1 |
|
Tube2 |
CD2 |
CD7 |
CD45 |
CD56 |
|
Tube3 |
CD8 |
CD3 |
CD45 |
CD4 |
|
Tube4 |
CD10 |
CD20 |
CD45 |
Blank |
|
Tubes |
CD5 |
CD11c |
CD45 |
CD19 |
|
Tube6 |
FMC7 |
CD23 |
CD45 |
CD20 |
|
Tube7 |
sKappa |
slambda |
CD45 |
CD19 |
|
Tube8 |
CD57 |
CD16 |
CD45 |
Blank |
- Lima M, Almeida J, Dos Anjos Teixeira M, AlgueroMdMdel C, Santos AH, Balanzategui A. et al. TCR +/CD4 + large granular lymphocytosis: A new clonal T-cell lymphoproliferative disorder. Am J Pathol 2003; 163: 763-71
- Olteanu H, Karandikar NJ, Eshoa C, Kroft SH. Laboratory findings in CD4 (+) large granular lymphocytoses. Int J Lab Hematol 2010; 32: e9-16
- Deepti M, Sharma RK, Kotru M, Saxena R. CD4+/CD8-/CD56+ T cell large granular lymphocyte proliferations; clonal disorders of uncertain significance. BMC Hematology 2014; 14: 9
- Rabade N, Mansukhani D, Khodaiji S, Padte B, Bhave A, Tembhare P. et al. Unusual immunophenotype of T-cell large granular lymphocytic leukemia: Report of two cases. Indian J PatholMicrobiol 2015; 58: 108-12
- Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. et al. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood 2004; 104: 328-35
- McKenna RW, Parkin J, Kersey JH, Gajl-Peczalska KJ, Peterson L, Brunning RD. Chronic lymphoproliferative disorder with unusualclinical, morphologic, ultrastructural and membrane surface markercharacteristics. Am J Med 1977; 62: 588-96
- Swerdlow SH, Campo E, Harris NL, Pileri SA, Stein H, Thiele J. et al. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. Lyon: IARC; 2008
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9: 247-58
Address for correspondence
Publication History
Received: 19 January 2018
Accepted: 27 April 2018
Article published online:
03 June 2021
© 2019. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1 The cells showed positivity for CD3, CD4, CD5, CD2, CD16, CD56, and - CD57, indicating a T-cell origin. There was aberrant loss of CD7 expression, and CD8 expression was negative
| Figure 2 Giemsa stained peripheral blood film (×100) showing large granular lymphocytes
- Lima M, Almeida J, Dos Anjos Teixeira M, AlgueroMdMdel C, Santos AH, Balanzategui A. et al. TCR +/CD4 + large granular lymphocytosis: A new clonal T-cell lymphoproliferative disorder. Am J Pathol 2003; 163: 763-71
- Olteanu H, Karandikar NJ, Eshoa C, Kroft SH. Laboratory findings in CD4 (+) large granular lymphocytoses. Int J Lab Hematol 2010; 32: e9-16
- Deepti M, Sharma RK, Kotru M, Saxena R. CD4+/CD8-/CD56+ T cell large granular lymphocyte proliferations; clonal disorders of uncertain significance. BMC Hematology 2014; 14: 9
- Rabade N, Mansukhani D, Khodaiji S, Padte B, Bhave A, Tembhare P. et al. Unusual immunophenotype of T-cell large granular lymphocytic leukemia: Report of two cases. Indian J PatholMicrobiol 2015; 58: 108-12
- Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. et al. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood 2004; 104: 328-35
- McKenna RW, Parkin J, Kersey JH, Gajl-Peczalska KJ, Peterson L, Brunning RD. Chronic lymphoproliferative disorder with unusualclinical, morphologic, ultrastructural and membrane surface markercharacteristics. Am J Med 1977; 62: 588-96
- Swerdlow SH, Campo E, Harris NL, Pileri SA, Stein H, Thiele J. et al. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. Lyon: IARC; 2008
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9: 247-58