 PDF
PDF  Views
Views  Share
Share


An Unusual Case of Acute Myeloid Leukemia with t(8:21) Presenting with Hypereosinophilia Showing Dysplastic Features
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(04): 612-614
DOI: DOI: 10.4103/ijmpo.ijmpo_197_19
Abstract
Acute myeloid leukemia (AML) with specific genetic abnormalities is a clinically, biologically, and prognostically distinct category with some of the entities in it displaying characteristic morphology. AML with t(8:21) is one such subtype carrying favorable prognosis with specific blast morphology. Eosinophilia, characteristically, has been described till date in AML with inv (16); however, hypereosinophilia with prominent dysplastic features has yet not been seen with any AML subtype. We report the case of an 8-year-old child presenting with massive splenomegaly, hypereosinophilia, and low marrow blast percentage. The initial clinical and hematological impression was that of a chronic myeloproliferative neoplasm, which was later diagnosed as AML with t(8:21) with the help of cytogenetic studies. The case report highlights the unusual and extremely rare presentation of this AML subtype and the importance of cytogenetic studies in definite categorization, especially in cases with overlapping morphological and immunophenotypic findings.
Publication History
Received: 14 September 2019
Accepted: 20 October 2019
Article published online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Acute myeloid leukemia (AML) with specific genetic abnormalities is a clinically, biologically, and prognostically distinct category with some of the entities in it displaying characteristic morphology. AML with t(8:21) is one such subtype carrying favorable prognosis with specific blast morphology. Eosinophilia, characteristically, has been described till date in AML with inv (16); however, hypereosinophilia with prominent dysplastic features has yet not been seen with any AML subtype. We report the case of an 8-year-old child presenting with massive splenomegaly, hypereosinophilia, and low marrow blast percentage. The initial clinical and hematological impression was that of a chronic myeloproliferative neoplasm, which was later diagnosed as AML with t(8:21) with the help of cytogenetic studies. The case report highlights the unusual and extremely rare presentation of this AML subtype and the importance of cytogenetic studies in definite categorization, especially in cases with overlapping morphological and immunophenotypic findings.
Introduction
Acute myeloid leukemia (AML) results from clonal proliferation of undifferentiated myeloid precursors leading to bone marrow failure. Various subsets of AML have been defined depending on the characteristic cytogenetic abnormality, which have prognostic implications. One such subset is AML with t(8:21) (q22:q22), which has a favorable prognosis and distinct biological characteristics.[1] It was the first cytogenetic abnormality detected in AML.[1] The translocation involves RUNX1 gene present on the chromosome 21 and RUNX1T1 gene on chromosome 8. This abnormality is found in 5%–10% of all AML cases and is most commonly seen in younger patients.[1] Morphologically, this category has characteristic blasts and increased numbers of neutrophils, eosinophils, and their precursors to an extent that the picture sometimes resembles that of a chronic myeloproliferative neoplasm, especially when the blast counts are low.[1] However, AML with t(8:21) presenting initially with massive splenomegaly and hypereosinophilia with many dysplastic eosinophils and their precursors is extremely rare. Hypereosinophilia in itself enlists many differential diagnoses and is an alarming finding in rapid turnover hematological states. It can be found either as an associated finding in acute leukemia or as a clonal proliferation in chronic eosinophilic leukemia. The dilemma might not always be entirely solved on morphology alone.
Case Report
An 8-year-old girl presented with fever for 3 months, pain in the abdomen, and progressive abdominal distension for 1 month. Fever was intermittent, high grade, and was not associated with vomiting. There was a history of weight loss, loss of appetite, and generalized weakness. There was no breathing difficulty, bleeding from orifices, or loose stools. On general examination, her general condition was poor. She was febrile and pale. Few petechial spots could be identified over the abdomen. There was no lymphadenopathy or icterus. On local examination, there was massive splenomegaly (10 cm below the costal margin) and mild hepatomegaly (1 cm below costal margin).
Hemogram showed hemoglobin level of 8.4 g/dl and a total leukocyte count of 120,000/ cubic mm with eosinophils accounting for 52%. Peripheral blood film (PBF) examination showed marked leukocytosis with striking predominance of eosinophils and their precursors, including promyelocytes, metamyelocytes, and myelocytes along with few blasts. Eosinophils and their precursors showed marked morphological abnormalities in the form of nuclear hyperlobation, coarse granules, and cytoplasmic vacoulations [Figure 1]a]. Platelets were markedly reduced (10,000/ cubic mm). Myeloperoxidase cytochemistry was strongly positive. Bone marrow biopsy was obtained, which showed a hypercellular marrow with marked infiltration by eosinophilic precursors in interstitial and paratrabecular pattern along with few immature precursor cells which were highlighted by immunohistochemistry for CD34 and CD117. Megakaryocytes were markedly reduced [Figure 1]b], [Figure 1]c], [Figure 1]d].

| Figure. 1 (a) Peripheral blood smear shows eosinophils with dysplastic features such as nuclear hyperlobation and coarse granules. Inset shows the presence of blasts (Giemsa, ×1000). (b and c) Diffuse replacement of bone marrow by eosinophils and their precursors (H and E ×1000, 100 respectively). (d) Bone marrow blasts showing positivity for CD34 (Immunohistochemistry, ×1000)
Flow cytometry was performed on peripheral blood. Cells were gated on CD45 versus side scatter. There were 17% blasts and 65% granulocytes (eosinophils and its precursors). The predominant population in eosinophils and precursors were positive for CD34, CD117, HLA-DR, and CD25 [Figure 2]a], [Figure 2]b], [Figure 2]c], [Figure 2]d], [Figure 2]e]. Based on the morphology and flow cytometry, showing predominant expression of variety of immature markers over the eosinophils and its precursors (reflecting acute neoplastic nature), the diagnosis considered was eosinophilic leukemia, and the possibility of AML M4E0 was suggested. Cytogenetic studies were advised to confirm the same and rule out other leukemic conditions associated with eosinophilia. Cytogenetic evaluation for PDGFR alpha, PDGFR beta, FGFR1 rearrangements, BCR-ABL, and inv(16) was negative. However, t(8:21) was positive in our case [Figure 2]f]. Thereafter, in conjunction with genetics, a final diagnosis of AML with t(8:21) with hypereosinophilia was given. The patient was initially being treated with hydroxyurea and steroids for symptomatic relief and now has been put up on therapy for AML, for which she responded well.
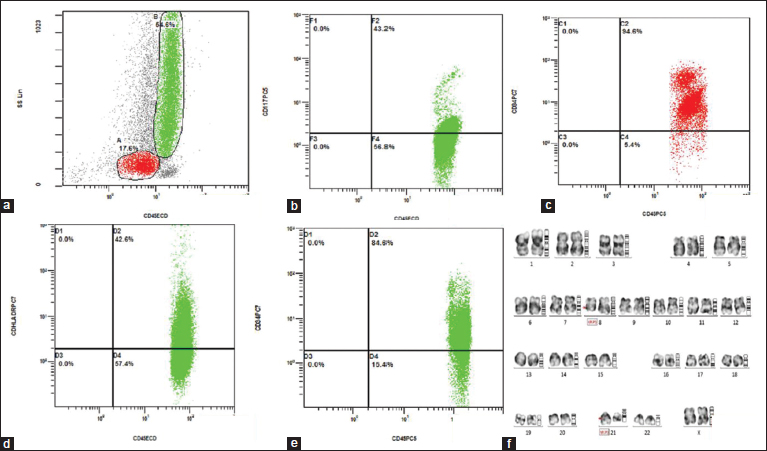
| Figure. 2(a) CD45 versus side scatter showing distinct populations of blasts (17%) and granulocytes (55%). (b,d,e) The gated granulocytic population with increased expression of HLA DR (42.6%), CD117 (43.2%), and CD34 (84.6%), respectively. (c) The gated blast population showing CD34 [removed]94.6%). (f) Chromosomal analysis: G-bands by trypsin using Giemsa banding showing reciprocal translocation between the chromosomes 8 and 21, involving the region q22 and q22, respectively (arrow)
Discussion
At present, AML with specific genetic abnormalities has been classified by the WHO under the broad category of “AML with recurrent genetic abnormalities.” These entities are unique and are clinically, biologically, as well as prognostically distinct and hence it is important to diagnose them. AML with t(8:21) is one such entity which shows good response to the conventional chemotherapy and thus has a favorable prognosis. It is also the most common chromosomal abnormality seen with AML. According to the current WHO criteria, the diagnosis of AML can be made even when the blast percentage (peripheral blood/bone marrow) is <20 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_197_19#JR_1" xss="removed">>1] Although specific morphological characteristics have been discussed with all these entities, sometimes variations do occur and can lead to a diagnostic dilemma. This type of AML has characteristic blast morphology and can be seen associated with bone marrow eosinophilia.[1] We described the case of a girl where the presence of massive splenomegaly and great morphological resemblance to eosinophilic leukemia on peripheral blood examination with blast count <20 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_197_19#JR_2" xss="removed">2] These are the cases where cytogenetic evaluation has immense decisive role and is the cornerstone to final diagnosis, as the etiology of eosinophilia is varied, encompassing both lesions which are benign/reactive and those which are out rightly malignant. Reactive causes of eosinophilia were ruled out in our case. Hematological malignancies linked with eosinophilia include CML, JMML, CEL, AML M4E0, AML with inv (16), ALL, myeloid, and lymphoid neoplasms associated with PDGFRA, PDGFRB, and FGFR1 rearrangements, which require both morphological and genetic support for diagnosis.
In our case, the karyotypic abnormality, t(8:21), was positive and a final decision of AML could be made even with low blast percentage. Immunophenotypically, the blast and the immature eosinophilic precursors both exhibited immature markers, including CD34, CD117, and HLA-DR. We also noticed increased CD25 expression, which has been described as a prognostic marker, associated with aggressive clinical behavior.
Review of literature revealed very few reports available with similar presentation with hypereosinophilia, rashes, and abnormal eosinophilic precursors in the bone marrow.[3],[4] However, the presence of eosinophils and its precursors in extreme numbers simulating eosinophilic leukemia with prominent dysplastic features is extremely rare and is uncommon in AML with t(8:21). This case report highlights the still another type of morphology that can be seen with this karyotypic abnormality. The exact pathogenesis behind eosinophilia observed in these cases is still unknown. However, a few published reports have shown that these atypical eosinophils might be derived from the leukemic clone itself by increased expression of interleukin-5 receptors as demonstrated byin vitro studies.[5]
Due to a lack of much data on this, how well these patients respond to the conventional antileukemic therapy is unknown. AML with t(8:21) in general has a favorable prognosis; however, hypereosinophilia in itself can cause a lot of parenchymal damage to the lungs, heart, and gastrointestinal tract due to the liberated cytokines, leading to organ fibrosis and increased morbidity.[5]
Our patient received both steroids and antileukemic therapy with daunorubicin and cytarabine. Repeat PBF after the completion of the first cycle showed marked reduction in the total leukocyte counts including the absolute eosinophil numbers. The patient is also recovering and is afebrile with moderate decrease in the spleen size.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- Reikvam H, Hatfield KJ, Kittang AO, Hovland R. Bruserud Ø. Acute myeloid leukemia with the t (8;21) translocation: Clinical consequences and biological implications. J Biomed Biotechnol 2011; 2011: 104631
- Sangle NA, Perkins SL. Core-binding factor acute myeloid leukemia. Arch Pathol Lab Med 2011; 135: 1504-9
- Ong SY, Ho LP, Yeo PM, Ng HJ, Ang AL. Marked hypereosinophilia and rash as initial presentation of acute myeloid leukemia with t (8;21). Int J Hematol 2017; 106: 149-50
- Gupta O, Aggarwal R, Prasad R. Acute myeloid leukemia (AML-M2) with translocation (8;21) (q22;q22) and abnormal eosinophilic precursors in the bone marrow – A case report. Indian J Pediatr 2012; 79: 256-9
- Ema H, Kitano K, Suda T, Sato Y, Muroi K, Ohta M. et al. In vitro differentiation of leukemic cells to eosinophils in the presence of interleukin-5 in two cases of acute myeloid leukemia with the translocation (8;21)(q22;q22). Blood 1990; 75: 350-6
Address for correspondence
Publication History
Received: 14 September 2019
Accepted: 20 October 2019
Article published
online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301
UP, India
| Figure. 1 (a) Peripheral blood smear shows eosinophils with dysplastic features such as nuclear hyperlobation and coarse granules. Inset shows the presence of blasts (Giemsa, ×1000). (b and c) Diffuse replacement of bone marrow by eosinophils and their precursors (H and E ×1000, 100 respectively). (d) Bone marrow blasts showing positivity for CD34 (Immunohistochemistry, ×1000)
| Figure. 2(a) CD45 versus side scatter showing distinct populations of blasts (17%) and granulocytes (55%). (b,d,e) The gated granulocytic population with increased expression of HLA DR (42.6%), CD117 (43.2%), and CD34 (84.6%), respectively. (c) The gated blast population showing CD34 [removed]94.6%). (f) Chromosomal analysis: G-bands by trypsin using Giemsa banding showing reciprocal translocation between the chromosomes 8 and 21, involving the region q22 and q22, respectively (arrow)
References
- Reikvam H, Hatfield KJ, Kittang AO, Hovland R. Bruserud Ø. Acute myeloid leukemia with the t (8;21) translocation: Clinical consequences and biological implications. J Biomed Biotechnol 2011; 2011: 104631
- Sangle NA, Perkins SL. Core-binding factor acute myeloid leukemia. Arch Pathol Lab Med 2011; 135: 1504-9
- Ong SY, Ho LP, Yeo PM, Ng HJ, Ang AL. Marked hypereosinophilia and rash as initial presentation of acute myeloid leukemia with t (8;21). Int J Hematol 2017; 106: 149-50
- Gupta O, Aggarwal R, Prasad R. Acute myeloid leukemia (AML-M2) with translocation (8;21) (q22;q22) and abnormal eosinophilic precursors in the bone marrow – A case report. Indian J Pediatr 2012; 79: 256-9
- Ema H, Kitano K, Suda T, Sato Y, Muroi K, Ohta M. et al. In vitro differentiation of leukemic cells to eosinophils in the presence of interleukin-5 in two cases of acute myeloid leukemia with the translocation (8;21)(q22;q22). Blood 1990; 75: 350-6