 PDF
PDF  Views
Views  Share
Share


Adrenal Tumors in Children: Spectrum of Presentation and Surgical Approach in a Tertiary Care Institute
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(03): 351-357
DOI: DOI: 10.4103/ijmpo.ijmpo_176_18
Abstract
Context: Adrenal tumors can arise either from cortex or from medulla; both the regions being structurally and functionally different. Current knowledge on childhood adrenocortical tumors (ACTs), the management approach, and the outcome is limited due to limited number of cases and studies. Neuroblastoma is a neoplasm of the neural crest origin and 50% arise from adrenal medulla. Pheochromocytoma (PCC) is an exciting and rare neoplasm in childhood. Aims: To evaluate the clinical presentation, spectrum of pathology, and management of adrenal tumors in children. Subjects and Methods: The study was conducted in a tertiary care pediatric institute over 5 years on children with intra-abdominal solid tumors. Adrenal tumors were diagnosed on radiological studies and postoperatively confirmed by histopathology. Results: Sixty-three patients were operated for intra-abdominal solid tumors, and Wilms tumor (39) was the most common finding. Of 11 cases of adrenal tumors confirmed by histopathology, eight were adrenal neuroblastoma, one 14-month-old female baby was detected as PCC, while two female children presented with the features of Cushing syndrome and virilization at the age of 3 and 7 years. Postoperatively, one of them was found to be adrenal adenoma and one was found to be adrenocortical carcinoma. Conclusions: ACTs are rare and potentially fatal in childhood. Complete surgical resection is the mainstay of therapy in adrenal tumors and is curative for associated hypertension. Neuroblastoma was the third common intra-abdominal solid tumor after Wilms tumor and retroperitoneal teratoma. The patient with PCC contributed to “Rule of 10s” because of the absence of hypertension and childhood occurrence.
Keywords
Adrenal adenoma - adrenocortical carcinoma - neuroblastoma - pheochromocytomaPublication History
Received: 12 August 2018
Accepted: 06 February 2020
Article published online:
28 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Context: Adrenal tumors can arise either from cortex or from medulla; both the regions being structurally and functionally different. Current knowledge on childhood adrenocortical tumors (ACTs), the management approach, and the outcome is limited due to limited number of cases and studies. Neuroblastoma is a neoplasm of the neural crest origin and 50% arise from adrenal medulla. Pheochromocytoma (PCC) is an exciting and rare neoplasm in childhood. Aims: To evaluate the clinical presentation, spectrum of pathology, and management of adrenal tumors in children. Subjects and Methods: The study was conducted in a tertiary care pediatric institute over 5 years on children with intra-abdominal solid tumors. Adrenal tumors were diagnosed on radiological studies and postoperatively confirmed by histopathology. Results: Sixty-three patients were operated for intra-abdominal solid tumors, and Wilms tumor (39) was the most common finding. Of 11 cases of adrenal tumors confirmed by histopathology, eight were adrenal neuroblastoma, one 14-month-old female baby was detected as PCC, while two female children presented with the features of Cushing syndrome and virilization at the age of 3 and 7 years. Postoperatively, one of them was found to be adrenal adenoma and one was found to be adrenocortical carcinoma. Conclusions: ACTs are rare and potentially fatal in childhood. Complete surgical resection is the mainstay of therapy in adrenal tumors and is curative for associated hypertension. Neuroblastoma was the third common intra-abdominal solid tumor after Wilms tumor and retroperitoneal teratoma. The patient with PCC contributed to “Rule of 10s” because of the absence of hypertension and childhood occurrence.
Keywords
Adrenal adenoma - adrenocortical carcinoma - neuroblastoma - pheochromocytomaIntroduction
Tumors arising from the adrenal cortex are usually adrenal adenomas or adrenocortical carcinomas (ACCs); whereas those arising from the adrenal medulla are neuroblastomas and pheochromocytomas (PCCs). Conventionally, they are classified into functional and nonfunctional tumors. Adrenal adenomas, ACCs, and PCCs are functional tumors, whereas neuroblastomas are nonfunctional. Adrenocortical tumors (ACTs) are rare in children, comprising <0.2% of pediatric neoplasms.[1]
Current knowledge on childhood ACT, the management approach, and the outcome is limited due to limited number of cases and studies.[2] Neuroblastoma is a neoplasm of the neural crest origin. Half of the reported cases of neuroblastoma arise from adrenal medulla, while the rest originate from paraspinal sympathetic ganglia in the retroperitoneum, posterior mediastinum, and pelvic organ of Zuckerkandl. PCC is an exciting and rare neoplasm in childhood. The term pheochromocytoma is used for catecholamine-secreting tumors originating in the adrenal medulla, whereas paraganglioma is the term used for extra-adrenal locations, such as sympathetic and parasympathetic paraganglia.[3] In spite of rarity, PCC accounts for 1% of hypertension in children.[4] PCC is well recognized for its 10% extra-adrenal location, 10% bilaterality, 10% malignant, 10% presentation in children and 10% cases without hypertension; “Rule of 10s”.[5] Therefore, this study was conducted to evaluate the clinical presentation, spectrum of pathology, and management of adrenal tumors in children.
Subjects and Methods
This retrospective study of data analysis was conducted in the department of pediatric surgery at our Institute. It was conducted over 5 years from January 2013 to December 2017. Patients with intra-abdominal solid tumors were observed, especially those over renal/adrenal locations. Inclusion criteria were (1) patients below the age of 14 years, (2) tumors arising from adrenal gland. Exclusion criteria were (1) patients above the age of 14 years, (2) patients with recurrent abdominal tumor. Detailed demographic data, family history of cancer, and weight and general condition on admission were recorded. General examination in the form of pallor, icterus, pedal edema, pulse, and blood pressure was done. Adrenal tumors were provisionally diagnosed on the basis of radiological studies; ultrasonography and computed tomography (CT) scan of the abdomen.
Routine hemogram, electrolytes, urea, creatinine, and liver function tests were done. Special biochemical investigations included estimation of serum cortisol, testosterone, and adrenocorticotrophic hormone (ACTH). Twenty-four-hour urinary catecolamines, homovanillic acid (HVA), and vanillylmandelic acid (VMA) were estimated. After establishing the provisional diagnosis, correction of general condition and anemia was done. Details about the disease, plan of management, and risks were explained to the parents. After written consent and preanesthetic checkup, surgery was undertaken under general anesthesia. Excision of the tumor was performed and the mass was sent for histopathological study along with immunohistochemistry. After operative recovery, oral diet and medications were started. Further advice was given according to the histopathology report and followed.
Statistical methods
Data are expressed in numbers and percentages. Confidence interval (CI) was used to express the range of values. Fisher's exact test 2 was used for comparison of various groups. P < 0.05 was considered statistically significant. Ethical considerations: This study was conducted in accordance with ethical standards and with the revised Declaration of Helsinki 2000.
Results
Sixty-three patients were operated for intra-abdominal solid tumors, and Wilms' tumor (39) was the most common pathology followed by retroperitoneal teratoma and adrenal tumors, 11 cases each [Table 1]. Out of 11 cases of adrenal tumors confirmed by histopathology, eight cases of adrenal neuroblastoma, one case of PCC, one case of adrenal adenoma, and one case of ACC were found [Table 2]. Although we included children below the age of 14 years, the maximum age of presentation of adrenal tumors was 7 years. The age range was from 7 to 72 months (CI 16.89–49.5) and the weight range was from 7.3 to 23.15 kg (CI 3.42–8.60). Four children (37%) were below the age of 1 year.{Table 1}{Table 2}
Intra-abdominal solid tumors operated during the study period
|
Pathology |
Number of cases |
|---|---|
|
Wilms tumor |
39 |
|
Retroperitoneal teratoma |
11 |
|
Adrenal tumor |
11 |
|
Rhabdomyosarcoma bladder |
1 |
|
Ovarian tumor |
1 |
|
Total |
63 |
The mean age of presentation of neuroblastoma cases was 29 months and the median age was 1 year and 3 months. There were six male and two female patients, with the male-to-female ratio being 3:1. A male preponderance was found in our study among neuroblastoma patients in comparison to other abdominal tumors (P = 0.26; Fisher's exact test 2). Family history of malignancy was not reported in any case. Abdominal mass and distension were the principal presenting features in all the children. Five patients complained of abdominal pain. There was recent loss of weight in all patients, and diarrhea was reported in two cases. Hypertension was detected in another two cases, which settled with antihypertensive drugs. The weight of the children ranged from 7.3 to 17.3 kg, with the mean weight being 11.78 kg. On physical examination, anemia was found in all cases. Ultrasonography and abdominal X-ray suggested mass lesions in all cases, but the provisional diagnosis was made on the basis of contrast-enhancing CT scan [Figure 1]a and [Figure 1]b. The tumor was found on the right side in four cases and the left side in four cases. Laparotomy and resection were done in all cases. Histopathological examination of the excised tumors revealed neuroblastoma in eight cases [Figure 1]c and d]. Immunohistochemistry in these pediatric small round cell tumors was confirmed as neuroblastoma by revealing neuron-specific enolase (NSE) positivity [Figure 1]e. It was a necessity to exclude other small round cell tumors by immunohistochemistry. The patients were given chemotherapy at a regional cancer institute and the follow-up continued.
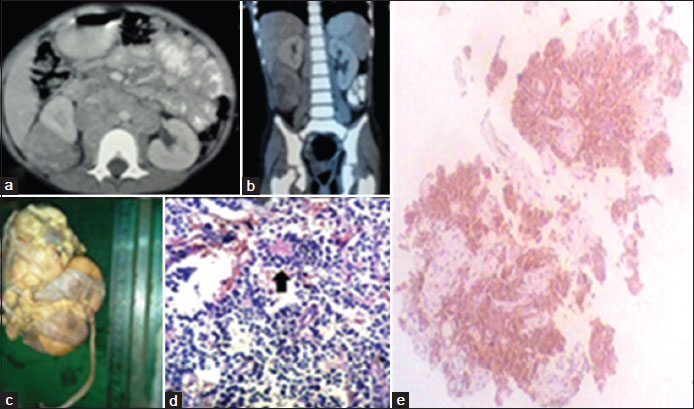
| Figure 1: A 6-year-old female child with right-sided adrenal tumor. (a and b) Contrast-enhancing computed tomography showing heterogeneous enhancing lesion in right lumbar region with areas of necrosis. (c) Gross pathology: Large soft, grayish relatively well circumscribed mass located above the upper pole of kidney. (d) Microsection: Small round tumor cells with typical homer wright rosette (black arrow), suggestive of neuroblastoma. (e) Positive immunohistochemistry marker for neuron-specific enolase in neuroblastoma
Two patients presented with the features of Cushing syndrome. Both of them were female children aged 3 and 7 years. They presented with moon facies; rounded face with prominent cheeks [Figure 2]a; acne, centripetal distribution of fat, and thin striae over the abdomen. The weights of the patients were 13 and 23.15 kg. The parents noticed recent weight gain, generalized obesity, weakness, and lethargy among these children. Hypertension was marked in both of them on admission; 134/88 and 130/90 mmHg, respectively. Features of virilization in the form of facial hair, pubic hair, and clitoral hypertrophy were well marked in both the children. Abdomen were soft, nontender, and without any palpable mass. Hemogram, serum urea, creatinine, liver function test, and coagulation profile were normal in both, but serum potassium was 2.08 mmol/l in the first child. Serum testosterone was raised in the first child (203 ng/dl), with reference value being 2–10 ng/dl along with dehydroepiandrosterone sulfate (DHEA-S, 710 mcg/dl), with reference value being 32–276. Plasma ACTH was normal in both the children. Twenty-four-hour urinary cortisol was raised in both the children, 639 and 870 mcg, respectively, with reference value being 28.5–213.7 mcg. However, 24-h urinary VMA and HVA were normal in both the patients along with normal metanephrine-creatinine ratio and normetanephrine-creatinine ratio.
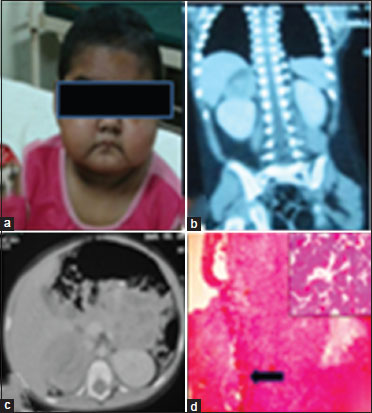
| Figure 2: A 3-year-old female child with right sided adrenocortical carcinoma. (a) Showing features of Cushing syndrome and virilization; moon facies, acne, and facial hair. (b and c) Contrast-enhanced computed tomography revealing exophytic heterogeneously enhancing mass lesion at the upper pole of the right kidney. (d) Tumor cells in diffuse architecture and shows capsular invasion (black arrow). (Inset) High nuclear grade with vascular invasion suggestive of adrenocortical carcinoma
Ultrasonography of the abdomen in the first patient suggested right adrenal mass, and CT scan revealed an exophytic heterogeneously enhancing mass lesion arising from the upper pole of the right kidney and infiltrating into the right renal vein [Figure 2]b and c]. Ultrasonography in the second patient suggested a mass in the region of left suprarenal gland, and CT scan revealed the presence of a globular solid lesion in the left adrenal measuring 18 mm × 20 mm [Figure 3]a. Patients were managed by multispecialty consultation, especially for the control of hypertension. However, the 3-year child suffered an attack of hypertensive encephalopathy during the preoperative period. The blood pressure recorded at that time was 180/110 mmHg but recovered after intensive care. She also needed potassium and calcium supplementation. After control of hypertension, the patients were operated under general anesthesia. Right-sided adrenalectomy was performed in the first patient, and left-sided adrenalectomy was done on the second patient [Figure 3]b and [Figure 3]c. Both the patients needed intensive care postoperatively. The first patient developed wound infection, which was controlled by regular dressing. The hypertension gradually decreased, and both the children became normotensive after surgery. Microscopy of the tumor mass in the first case showed diffuse architecture of growth pattern, with both venous and capsular invasions. Clear cell components constituted <25% with areas of myxoid change and necrosis. Higher power depicted high nuclear grade with mitosis >5/50 high-power fields (HPF). The features satisfied modified Weiss criteria for the diagnosis of ACC [Figure 2]d. Histopathology in the second case revealed adrenal adenoma [Figure 3]d.{Figure 3}
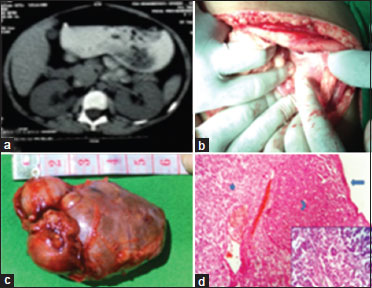
| Figure 3: A 7-year-old female child with left sided adrenocortical adenoma. (a) Computed tomography scan showing left adrenal lesion. (b) Intraoperative photograph of adrenal tumor. (c) Excised specimen. (d) Encapsulated tumor mass; capsule free from tumor cell infiltration (long arrow). Cells resembling zona glomerulosa (eosinophilic cytoplasm; arrow head) and fasciculata (vacuolated cytoplasm; short arrow) in solid sheets separated by sinusoidal blood vessels (Inset), suggestive of adrenocortical adenoma
One patient of adrenal tumor was found to have PCC. This case was a 14-month-old female, who presented as abdominal mass predominantly in the right hypochondrium extending to the lumbar region. The mass was firm and nontender and had an irregular surface. One important feature was that the child was normotensive. Ultrasonography suggested a heterogeneous solid mass measuring 9 cm × 7 cm with cystic components. CT scan detected a heterogeneous enhancing lesion measuring 7.3 cm × 8.0 cm × 9.1 cm in the upper pole of right kidney with foci of internal calcification [Figure 4]a and [Figure 4]b. She had urinary VMA of 1.70 mg/24 h, which was within normal limits. During surgery, the lesion was found to be adherent to the liver, portal vein, and inferior vena cava. It was an adrenal mass and separate from kidney. The tumor was removed without any spillage. Gross pathological examination found the lesion to be encapsulated measuring 9 cm × 8 cm × 7 cm. Cut-section showed grayish-yellow color, soft with areas of hemorrhage and necrosis. The microsection revealed tumor cells arranged in Zellballen and trabeculae pattern. Tumor cells had a basophilic-to-amphophillic cytoplasm with intracytoplasmic hyaline globules. The nuclei revealed prominent nucleoli having intracytoplasmic inclusions with salt and pepper chromatin at places [Figure 4]c and [Figure 4]d. The histopathological findings were consistent with adrenal PCC. The immunohistochemistry marker for chromogranin was positive confirming PCC [Figure 4]e. Although the size of the mass was 9 cm, it was considered benign because of insignificant mitotic rates and negative lymph node status. She is totally asymptomatic, gaining weight with normal investigations till 2-year follow-up.{Figure 4}
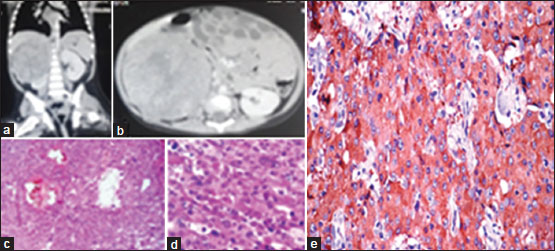
| Figure 4: A 1‑year 2‑month‑old female child with pheochromocytoma. (a and b) Computed tomography scan revealing 9.1 cm × 8 cm × 7.3 cm heterogeneous enhancing lesion near the upper pole of the right kidney, suggestive of right‑sided adrenal tumor. (c) Microsection; tumor cells arranged in Zellballen and trabeculae pattern with hemorrhage and cystic changes (H and E, ×100). (d) Pleomorphic tumor cells with granular amphophilic cytoplasm, intracytoplasmic inclusions, and prominent nucleoli, suggestive of pheochromocytoma. (e) Positive immunohistochemistry marker for chromogranin A confirming pheochromocytoma
Discussion
Neuroblastoma is the third most common cancer in children after leukemia and tumors of the central nervous system and is the most common among abdominal tumors.[6],[7] However, we found neuroblastoma as the third most common solid tumor in the abdomen after Wilms' tumor and retroperitoneal teratoma. Although neuroblastomas account for 8% of pediatric malignancies, it causes 15% of cancer deaths in children.[8] The median age of presentation is reported to be 22 months,[9] but in this study, it was earlier, i.e., 15 months. Males were more commonly affected in neuroblastoma as compared to other common abdominal tumors (P = 0.26) in our study [Table 2] and [Figure 5]. Adrenal neuroblastoma patients present with abdominal mass and pain. Loss of weight, appetite, and shortness of breath are usually associated and also observed in our study. Loose motion and diarrhea were found in 25% of our cases, settled with medical management. Diarrhea in neuroblastoma patients may be due to vasoactive intestinal peptide secretion by the tumor and sometimes may be severe and explosive in nature or associated with hypokalemia.[10] Anemia was a constant feature in our patients, and hypertension was found in 25% of our cases, settled with medical management. Diarrhea in neuroblastoma patients may be due to vasoactive intestinal peptide secretion by the tumor and sometimes may be severe and explosive in nature or associated with hypokalemia.[10] Anemia was a constant feature in our patients, and hypertension was found in 25% cases: The observations were similar to other studies. Hypertension is due to catecholamine production by the cancer cells. However, paraneoplastic syndromes, such as opsomyoclonus and nystagmus (dancing eye syndrome), reported in some series were not observed in our cases.[9] On ultrasonography, adrenal neuroblastomas appear as heterogeneous echogenic masses. CT scan is required for an accurate diagnosis, staging, and management. Origin of the tumor, vascular involvement, distant metastasis, and lymphadenopathy are clearly delineated. Areas of low attenuation inside the mass represent hemorrhage and necrosis. In spite of the microscopic diagnosis of excised tumors, it was a necessity to exclude other small round cell tumors by applying NSE as an immunohistochemistry marker. The features with stoma poor undifferentiated tumors presenting at more than 5 years of age carry an unfavorable course as per neuroblastoma grading system (Shimada). Cytogenetic setup is not available in study center. Moreover, it is not mandatory in such type of tumors because prognostication was fulfilled by Shimada criteria on morphology. Prognosis of children with advanced stage with distant metastasis is poor even after aggressive multimodality approach.[9]{Figure 5}
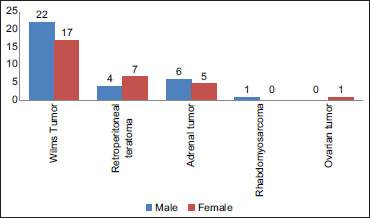
| Figure 5: Gender distribution of the study population
ACTs contribute 6% of pediatric adrenal tumors.[11] In our series, ACT was detected in two out of 11 cases of adrenal tumors. The incidence of ACC is reported to be 0.5/1 million children. However, in Brazil, the incidence is 1.5/1 million; three times more than the international figure, the reason being unclear.[12] Although the first case of ACT was reported in Lancet in 1865, most of the children with functioning ACT, who had undergone adrenalectomy till 1937, did not survive postoperatively due to adrenocortical insufficiency.[13] After the invent of cortisol, the prognosis improved. Vast majority (91.8%) of pediatric ACC occurs among Caucasians and Asians are affected only in 5.9% of cases.[14] To the best of the author's knowledge, ACC in this series is the only reported case among children in Odisha, having a population of 4.5 crores. Caucasian females are most commonly affected by ACCs, especially young children.[15],[16] A gender-specific change in the adrenal gland physiology is suggested for this female preponderance, but not conclusive. ACTs have a bimodal age distribution, with first and fifth decades of life being peaks.[17] Two genetic syndromes have a clear association with ACT: (i) Li–Fraumeni syndrome, a familial cancer syndrome having mutation of P53 gene, and (ii) Beckwith–Wiedmann syndrome having mutations in the 11p15 region.[1],[13] Genetic alterations or the above congenital syndromes are associated with about half of the cases of ACC.[18] About 90% of the cases of ACT are functional in children in contrast to <50 percent cases in adults.[19] Virilization is the most common presentation of ACT seen in 80%–84% of cases.[13],[19] It was present in both of our cases at an early age in the form of acne, pubic hair, and hypertrophy of clitoris. Cushing syndrome is the second most common presentation in childhood ACT seen in one-third cases with moon facies, centripetal distribution of fat, and striae over the skin and plethora.[13] Both of our cases of ACT were unique in that features of endocrine excess were manifested in them in the form of precocious puberty, virilization, and Cushing syndrome. Hypertension was also seen in both the cases and hypokalemia (feature of hyperaldosterinism) in one case. Hypertension is reported in 43% of cases of ACT.[7] Hypertensive crisis was seen in one of our case along with hypertensive encephalopathy.
Workup with hormonal estimation started in both these children as we suspected ACT. Free cortisol in 24-h urine was raised in both of them. This test is highly sensitive for Cushing syndrome. Plasma ACTH level was normal (ACTH-independent), suggesting tumors of adrenal origin. Twenty-four-hour urinary VMA, HVA, epinephrine, and norepinephrine were found to be normal in both the patients, provisionally excluding PCC, neuroblastoma, and other neural crest tumors. Serum testosterone and DHEA-S levels were raised in both the children. Serum DHEA-S is a specific test for androgen production and very specific for malignant tumors of the adrenal cortex.[20] The diagnosis of ACT was supported by the raised levels of urinary steroids, plasma testosterone, and DHEA-S in the presence of normal ACTH. Ultrasonography revealed an adrenal mass in both of our cases of ACT. CT scan can identify adrenal lesions measuring 0.5 cm. Although it is difficult to differentiate adenoma from carcinoma, features that favor ACC on CT scan are (1) tumor size: >4 cm (having a sensitivity of 93% for ACC), (2) shape: irregular, (3) texture: heterogeneous due to hemorrhage and necrosis, (4) attenuation: >10 Hounsfield unit; benign tumors are more lipid-rich and less dense, and (5) contrast washout: less than 40% after 15 min.[19],[21]
Surgery is the cornerstone of the treatment in ACTs. Our first patient needed radical surgical excision of the tumor, whereas the second patient required only adrenalectomy. Postoperative steroid replacement was given as both of our cases were having functional tumors. Functional ACTs are presumed to cause suppression of the contralateral adrenal gland.[13] Although the clinical presentation in both of our cases were similar, histopathology suggested ACC in the first case and adrenal adenoma in the second. Modified Weiss criteria is an established method for the diagnosis of ACC.[22],[23] Out of nine histological criteria, three findings are required to make the diagnosis of ACC[21] and our case fulfilled five Weiss criteria. The morphology was characteristic of ACC. Again, no single immunohistochemistry marker is 100% sensitive or specific in ACC. Although combination chemotherapy with etoposide and cisplatin was described in the postoperative management, the role of chemotherapy is not yet established due to limited number of cases.[1],[24] Adjuvant therapy with mitotane (dichlorodiphenyldichloroethane) is an established option in advanced and metastatic ACC in adults.[2] This is an insecticide derivative causing adrenocortical necrosis and tumor regression. However, its role in children pediatric ACC is not well established.[24] Chemotherapy was needed in our patients, and they were effectively managed by surgery alone. Age at diagnosis is considered as an important prognostic factor, with children <2 years having good outcome. Children >2 years of age are reported to be having only 29% survival rate.[25] However, both of our patients are disease-free and under regular follow-up. Microscopically, mitotic counts >20/50 HPF are described to be associated with poor outcome.[26] Recently, Ki-67 proliferation index has been identified as the most important prognostic factor for recurrence-free survival.[27]
PCC is a rare neoplasm arising from chromaffin cells of the adrenal medulla. Although the mean age of presentation reported in a recent study is 13.2 years,[28] it can present at a very young age as in our case (13 months). Hypertension is the most common presentation in pediatric PCC and is sustained in 90% of cases.[5],[25] However, childhood PCC may present without hypertension and without a classic triad[3] of headache, palpitation, and diaphoresis as in our study. Surgical excision is the treatment of PCC and leads to cure of hypertension.[29]
Management of adrenal tumors in children is challenging and needs multidisciplinary approach. Childhood hypertension due to adrenal tumor is severe and potentially a fatal condition. The extreme rarity of ACTs in our part of world as compared to some other regions, especially Southern Brazil, may point toward the role of environmental and genetic factors. Surgery is the optimal treatment and an event-free survival is expected even if the age group falls within poor prognostic criteria. Although neuroblastoma is reported as the most common abdominal tumor in childhood, it was found to be the third most common neoplasm after Wilms' tumor and retroperitoneal teratoma. However, there is a trend for earlier age at presentation as compared to previous reports. PCC detected and managed in our study is not only a rare finding but also contributed to the “Rule of 10s” due to very younger age at presentation and absence of hypertension. Our study shows that complete surgical resection is the mainstay of therapy in adrenal tumors and is curative for associated hypertension.
Conflict of Interest
There are no conflicts of interest.
References
- Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: The mainstay of treatment. J Pediatr Surg 2004; 39: 759-63
- Mahendraraj K, Lau CS, Sidhu K, Chamberlain RS. Adrenocortical carcinoma in adults and children: A population-based outcomes study involving 1,623 patients from the surveillance, epidemiology, and end result (SEER) database (1973-2012). Clin Surg 2016; 1: 1017-7
- Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M. et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2010; 95: 2023-37
- Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C. et al. Phaeochromocytoma in children. Arch Dis Child 2008; 93: 899-904
- Tripathy PK, Pattnaik K, Nayak M, Mohanty HK. Pheochromocytoma in a child without hypertension: A contribution to the “Rule of 10s”. Indian J Med Paediatr Oncol 2017; 38: 59-61
- Kushner BH. Neuroblastoma: A disease requiring a multitude of imaging studies. J Nucl Med 2004; 45: 1172-88
- McHugh K. Renal and adrenal tumours in children. Cancer Imaging 2007; 7: 41-51
- Hiorns MP, Owens CM. Radiology of neuroblastoma in children. Eur Radiol 2001; 11: 2071-81
- Bittman ME, Lee EY, Restrepo R, Eisenberg RL. Focal adrenal lesions in pediatric patients. AJR Am J Roentgenol 2013; 200: W542-56
- Rich BS, Quaglia MP. Neuroblastoma. In: Coran AG, Adzick NS, Krummel TM, Laberge J, Shamberger RC. editors Pediatric Surgery. Philadelphia: Elsevier Saunders; 2012. 7. 441-58
- Chudler RM, Kay R. Adrenocortical carcinoma in children. Urol Clin North Am 1989; 16: 469-79
- Ribeiro RC, Sandrini Neto RS, Schell MJ, Lacerda L, Sambaio GA, Cat I. Adrenocortical carcinoma in children: A study of 40 cases. J Clin Oncol 1990; 8: 67-74
- Ribeiro RC, Michalkiewicz EL, Figueiredo BC, DeLacerda L, Sandrini F, Pianovsky MD. et al. Adrenocortical tumors in children. Braz J Med Biol Res 2000; 33: 1225-34
- McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: A surveillance, epidemiology, and end results (SEER) program study. J Pediatr Surg 2013; 48: 1025-31
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG. et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 2004; 22: 838-45
- Kerkhofs TM, Ettaieb MH, Verhoeven RH, Kaspers GJ, Tissing WJ, Loeffen J. et al. Adrenocortical carcinoma in children:First population-based clinicopathological study with long-term follow-up. Oncol Rep 2014; 32: 2836-44
- Liou LS, Kay R. Adrenocortical carcinoma in children. Review and recent innovations. Urol Clin North Am 2000; 27: 403-21
- Ribeiro RC, Fighueiredo B. Childhood adrenocortical tumors. Eur J Cancer 2004; 40: 1117-26
- Gundgurthi A, Kharb S, Dutta MK, Garg MK, Khare A, Jacob MJ. et al. Childhood adrenocortical carcinoma: Case report and review. Indian J Endocrinol Metab 2012; 16: 413-5
- Diesen DL, Skinner MA. et al. Endocrine disorders and tumors. In: Holcomb GW 3rd, Murphy JP, Ostlie DJ, editors. Ashcraft's Pediatric Surgery. Philadelphia: Elsevier Saunders 2014; 6: 1067-85
- Wanis KN, Kanthan R. Diagnostic and prognostic features in adrenocortical carcinoma: A single institution case series and review of the literature. World J Surg Oncol 2015; 13: 117
- Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol 2009; 40: 757-68
- Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C. et al. Weiss system revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol 2002; 26: 1612-9
- Cho MJ, Kim DY, Kim SC, Kim TH, Kim IK. Adrenocortical tumors in children 18 years old and younger. J Korean Surg Soc 2012; 82: 246-50
- Ciftci AO, Senocak ME, Tanyel FC, Büyükpamukçu N. Adrenocortical tumors in children. J Pediatr Surg 2001; 36: 549-54
- Libé R. Adrenocortical carcinoma (ACC): Diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015; 3: 45
- Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F. et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 2015; 100: 841-9
- Eren E, Saglam H, Caliskan Y, Kiristioglu I, Tarim O, Sarin YK. Pediatric patients with pheochromocytoma: Experience of a tertiary health center. Pediatr Int 2015; 57: 875-9
- Mishra A, Mehrotra PK, Agarwal G, Agarwal A, Mishra SK. Pediatric and adolescent pheochromocytoma: Clinical presentation and outcome of surgery. Indian Pediatr 2014; 51: 299-302
Address for correspondence
Publication History
Received: 12 August 2018
Accepted: 06 February 2020
Article published online:
28 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: A 6-year-old female child with right-sided adrenal tumor. (a and b) Contrast-enhancing computed tomography showing heterogeneous enhancing lesion in right lumbar region with areas of necrosis. (c) Gross pathology: Large soft, grayish relatively well circumscribed mass located above the upper pole of kidney. (d) Microsection: Small round tumor cells with typical homer wright rosette (black arrow), suggestive of neuroblastoma. (e) Positive immunohistochemistry marker for neuron-specific enolase in neuroblastoma
| Figure 2: A 3-year-old female child with right sided adrenocortical carcinoma. (a) Showing features of Cushing syndrome and virilization; moon facies, acne, and facial hair. (b and c) Contrast-enhanced computed tomography revealing exophytic heterogeneously enhancing mass lesion at the upper pole of the right kidney. (d) Tumor cells in diffuse architecture and shows capsular invasion (black arrow). (Inset) High nuclear grade with vascular invasion suggestive of adrenocortical carcinoma
| Figure 3: A 7-year-old female child with left sided adrenocortical adenoma. (a) Computed tomography scan showing left adrenal lesion. (b) Intraoperative photograph of adrenal tumor. (c) Excised specimen. (d) Encapsulated tumor mass; capsule free from tumor cell infiltration (long arrow). Cells resembling zona glomerulosa (eosinophilic cytoplasm; arrow head) and fasciculata (vacuolated cytoplasm; short arrow) in solid sheets separated by sinusoidal blood vessels (Inset), suggestive of adrenocortical adenoma
| Figure 4: A 1‑year 2‑month‑old female child with pheochromocytoma. (a and b) Computed tomography scan revealing 9.1 cm × 8 cm × 7.3 cm heterogeneous enhancing lesion near the upper pole of the right kidney, suggestive of right‑sided adrenal tumor. (c) Microsection; tumor cells arranged in Zellballen and trabeculae pattern with hemorrhage and cystic changes (H and E, ×100). (d) Pleomorphic tumor cells with granular amphophilic cytoplasm, intracytoplasmic inclusions, and prominent nucleoli, suggestive of pheochromocytoma. (e) Positive immunohistochemistry marker for chromogranin A confirming pheochromocytoma
| Figure 5: Gender distribution of the study population
References
- Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: The mainstay of treatment. J Pediatr Surg 2004; 39: 759-63
- Mahendraraj K, Lau CS, Sidhu K, Chamberlain RS. Adrenocortical carcinoma in adults and children: A population-based outcomes study involving 1,623 patients from the surveillance, epidemiology, and end result (SEER) database (1973-2012). Clin Surg 2016; 1: 1017-7
- Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M. et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2010; 95: 2023-37
- Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C. et al. Phaeochromocytoma in children. Arch Dis Child 2008; 93: 899-904
- Tripathy PK, Pattnaik K, Nayak M, Mohanty HK. Pheochromocytoma in a child without hypertension: A contribution to the “Rule of 10s”. Indian J Med Paediatr Oncol 2017; 38: 59-61
- Kushner BH. Neuroblastoma: A disease requiring a multitude of imaging studies. J Nucl Med 2004; 45: 1172-88
- McHugh K. Renal and adrenal tumours in children. Cancer Imaging 2007; 7: 41-51
- Hiorns MP, Owens CM. Radiology of neuroblastoma in children. Eur Radiol 2001; 11: 2071-81
- Bittman ME, Lee EY, Restrepo R, Eisenberg RL. Focal adrenal lesions in pediatric patients. AJR Am J Roentgenol 2013; 200: W542-56
- Rich BS, Quaglia MP. Neuroblastoma. In: Coran AG, Adzick NS, Krummel TM, Laberge J, Shamberger RC. editors Pediatric Surgery. Philadelphia: Elsevier Saunders; 2012. 7. 441-58
- Chudler RM, Kay R. Adrenocortical carcinoma in children. Urol Clin North Am 1989; 16: 469-79
- Ribeiro RC, Sandrini Neto RS, Schell MJ, Lacerda L, Sambaio GA, Cat I. Adrenocortical carcinoma in children: A study of 40 cases. J Clin Oncol 1990; 8: 67-74
- Ribeiro RC, Michalkiewicz EL, Figueiredo BC, DeLacerda L, Sandrini F, Pianovsky MD. et al. Adrenocortical tumors in children. Braz J Med Biol Res 2000; 33: 1225-34
- McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: A surveillance, epidemiology, and end results (SEER) program study. J Pediatr Surg 2013; 48: 1025-31
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG. et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 2004; 22: 838-45
- Kerkhofs TM, Ettaieb MH, Verhoeven RH, Kaspers GJ, Tissing WJ, Loeffen J. et al. Adrenocortical carcinoma in children:First population-based clinicopathological study with long-term follow-up. Oncol Rep 2014; 32: 2836-44
- Liou LS, Kay R. Adrenocortical carcinoma in children. Review and recent innovations. Urol Clin North Am 2000; 27: 403-21
- Ribeiro RC, Fighueiredo B. Childhood adrenocortical tumors. Eur J Cancer 2004; 40: 1117-26
- Gundgurthi A, Kharb S, Dutta MK, Garg MK, Khare A, Jacob MJ. et al. Childhood adrenocortical carcinoma: Case report and review. Indian J Endocrinol Metab 2012; 16: 413-5
- Diesen DL, Skinner MA. et al. Endocrine disorders and tumors. In: Holcomb GW 3rd, Murphy JP, Ostlie DJ, editors. Ashcraft's Pediatric Surgery. Philadelphia: Elsevier Saunders 2014; 6: 1067-85
- Wanis KN, Kanthan R. Diagnostic and prognostic features in adrenocortical carcinoma: A single institution case series and review of the literature. World J Surg Oncol 2015; 13: 117
- Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol 2009; 40: 757-68
- Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C. et al. Weiss system revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol 2002; 26: 1612-9
- Cho MJ, Kim DY, Kim SC, Kim TH, Kim IK. Adrenocortical tumors in children 18 years old and younger. J Korean Surg Soc 2012; 82: 246-50
- Ciftci AO, Senocak ME, Tanyel FC, Büyükpamukçu N. Adrenocortical tumors in children. J Pediatr Surg 2001; 36: 549-54
- Libé R. Adrenocortical carcinoma (ACC): Diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015; 3: 45
- Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F. et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 2015; 100: 841-9
- Eren E, Saglam H, Caliskan Y, Kiristioglu I, Tarim O, Sarin YK. Pediatric patients with pheochromocytoma: Experience of a tertiary health center. Pediatr Int 2015; 57: 875-9
- Mishra A, Mehrotra PK, Agarwal G, Agarwal A, Mishra SK. Pediatric and adolescent pheochromocytoma: Clinical presentation and outcome of surgery. Indian Pediatr 2014; 51: 299-302