 PDF
PDF  Views
Views  Share
Share


A Review on the Role of Molecular Genetics in the Diagnostic Workup of BCR::ABL1-Negative Myeloproliferative Neoplasms
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(06): 578-584
DOI: DOI: 10.1055/s-0043-1766138
Abstract
The diagnostic evaluation of myeloproliferative neoplasms (MPNs) depends on the close correlation between clinical features, morphologic assessment of a trephine bone marrow biopsy, and molecular markers. Typically, MPNs have driver mutations in JAK2, CALR, or MPL, as well as mutations in genes related to epigenetic regulation, RNA splicing, and signaling. Mutations in these genes are a hallmark of diagnostic, prognostic, and therapeutic assessment in patients with MPNs. In line with the World Health Organization classification, all myeloproliferative disorders require molecular characterization to support diagnoses or confirm entities defined by underlying molecular abnormalities. A structured molecular analysis workflow is essential for a rapid and cost-effective diagnosis of MPN. The purpose of this review is to explore the role of molecular diagnostics in the assessment of BCR::ABL1-negative MPNs.
Keywords
molecular diagnostics - MPN - PV - ET - PMF - non-classical MPNAuthors' Contributions
All the authors approve the submitted version of the manuscript. M.M. and A.K.A. wrote the paper. A.B.K., U.P.K., and B.P. contributed to the critical revisions of the manuscript.
Ethical Disclosure
None.
Supplementary MaterialPublication History
Article published online:
17 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
The diagnostic evaluation of myeloproliferative neoplasms (MPNs) depends on the close correlation between clinical features, morphologic assessment of a trephine bone marrow biopsy, and molecular markers. Typically, MPNs have driver mutations in JAK2, CALR, or MPL, as well as mutations in genes related to epigenetic regulation, RNA splicing, and signaling. Mutations in these genes are a hallmark of diagnostic, prognostic, and therapeutic assessment in patients with MPNs. In line with the World Health Organization classification, all myeloproliferative disorders require molecular characterization to support diagnoses or confirm entities defined by underlying molecular abnormalities. A structured molecular analysis workflow is essential for a rapid and cost-effective diagnosis of MPN. The purpose of this review is to explore the role of molecular diagnostics in the assessment of BCR::ABL1-negative MPNs.
Keywords
molecular diagnostics - MPN - PV - ET - PMF - non-classical MPNIntroduction
The myeloproliferative neoplasms (MPNs) are a group of related clonal hematologic disorders characterized by excess accumulation of one or more myeloid cell lineages. Symptoms include fatigue, pruritus, early satiety due to splenomegaly, increased infection risk, and thrombotic events. Thromboembolic and hemorrhagic events contribute to disease morbidity. There is a greater incidence of transformation to myelofibrosis with polycythemia vera (PV) than essential thrombocythemia (ET) and secondary acute myeloid leukemia (AML) with primary myelofibrosis (PMF) than with PV or ET. These are traditionally classified into four subgroups, which include (i) chronic myeloid leukemia (CML), (ii) classical Philadelphia-negative MPNs, that is, PV, ET, PMF; (iii) nonclassical Philadelphia-negative MPNs, that is, chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia (CEL); and (iv) MPN, not otherwise specified (NOS). Juvenile myelomonocytic leukemia (JMML) is now classified under BCR::ABL1-negative MPN of childhood as opposed to the earlier MDS/MPN category.[1] [2] [3]
The initial diagnostic evaluation of MPNs depends on a close correlation between clinical features such as splenomegaly, arterial or venous thrombosis, peripheral blood indices, morphologic assessment of a bone marrow biopsy, and molecular diagnostics. Increasing knowledge of molecular alterations has influenced paradigm shift in classification, which define entities based partially or exclusively on recurrent genetic mutations.[3] [4] Hence, an integrated multimodal approach is required for precise classification.
A mutation in codon 617 of JAK2, resulting in the replacement of the amino acid valine with phenylalanine [JAK2(NM_004972.3):c.1849G > T (p.Val617Phe)], is found in the vast majority of classical MPN cases. This gain of function mutation is present in approximately 96%-of PV, 55%-of ET, and 65%-of PMF.[5] JAK2 exon 12 mutations are described in 3-5%-of cases of PV and rarely in PMF.[5] [6] Thrombopoietin receptor (MPL) mutations have been reported to occur in 4 to 10%-of cases of JAK2-negative ET and PMF[7] [8] and calreticulin (CALR) mutations in 20 to 25%-of ET and PMF cases.[4] [9]
The triple-negative MPN is a molecular entity that lacks mutations in JAK2, CALR, or MPL genes but still exhibits histological and phenotypic features consistent with MPNs. Triple-negative cases of ET and PMF account for approximately 20%-and 5 to 10%-of cases, respectively.[10] [11] Molecular investigations should be expanded in this subtype of MPN patients to identify gene mutations that are not restricted to classical MPNs and to find evidence of disease clonality.[3] [12]
Nondriver somatic mutations implicated in the disease pathogenesis of MPNs belong to several functional classes. These include mutations in the epigenetic regulator genes such as DNA methylation and chromatin modifiers, genes involved in RNA splicing, signaling pathways, transcription factors, tumor suppressors, DNA damage response pathways, and cohesin complex. Among these, the most frequent nondriver mutations are found in the epigenetic regulator genes TET2, ASXL1, DNMT3A, and EZH2 and in the RNA splicing genes SRSF2 and U2AF1.[13] [14] [15] [16] [17] [18] [19] [20]
Diagnosing nonclassical MPNs (CNL, CEL, JMML) as well as MPN, NOS requires the exclusion of BCR::ABL1 fusion as well as molecular testing to screen for category-specific mutations such as CSF3R, RAS pathway genes, and other myeloid neoplasia-associated mutations.[2] [21]
Identifying molecular markers not only offers a simplified strategy for the diagnosis but also adds prognostic information that can guide management. Several genes, including ASXL1, EZH2, IDH1, IDH2, and SRSF2, are associated with shorter survival and increased risk of leukemic transformation (LT) in PMF patients.[22] These mutations define a higher risk category called “high molecular risk” (HMR). Molecular analysis may, therefore, aid in treatment decisions such as allogeneic stem cell transplantation.[22] [23]
This review aims to highlight the role of molecular diagnostics in each of these entities categorized under BCR::ABL1-negative MPN, as well as the need for a structured diagnostic testing algorithm in aiding rapid and cost-effective diagnosis.
Overview of BCR::ABL1-Negative Myeloproliferative Neoplasms and Screening Investigations
Polycythemia Vera
PV is a chronic MPN characterized by increased production of red blood cells independent of the erythropoiesis-regulating mechanisms. A somatic gain-of-function mutation JAK2V617F or other functionally similar JAK2 mutations that result in panmyelosis is detected in most patients with polycythemia vera. A diagnosis of PV is based on composite clinical, laboratory, and bone marrow histological features and requires the exclusion of all causes of secondary erythrocytosis, heritable polycythemia, and other MPNs[21] ([Table 1]).
|
Major criteria |
|
|---|---|
|
1 |
Elevated hemoglobin concentration (> 16.5 g/dL in men;> 16.0 g/dL in women) or Elevated hematocrit (>49%-in men; >48%-in women) or Increased red blood cell mass (> 25%-above mean normal predicted value) |
|
2 |
Bone marrow biopsy showing age-adjusted hypercellularity with trilineage growth (panmyelosis), including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size) |
|
3 |
Presence of JAK2 V617F or JAK2 exon 12 mutation |
|
Minor criteria |
|
|
Subnormal serum erythropoietin level |
|
Laboratory detection of JAK2V617F is highly sensitive (97%-sensitivity) and virtually 100%-specific for identifying PV from other causes of increased hematocrit. A subnormal level of erythropoietin (EPO) is expected in 85%-of PV patients. As a result of simultaneous measurement of serum EPO level, the possibility of false positive or false-negative mutation test results can be effectively avoided.[24]
JAK2-negative cases with persisting and significant erythrocytosis are classified as “JAK2-negative PV” only after all probable causes of secondary erythrocytosis have been excluded. As the British Society of Haematology (BSH) 2019 guidelines outlined, this rare entity must meet stringent criteria to differentiate from idiopathic and secondary erythrocytosis. The World Health Organization (WHO) criteria for the diagnosis of PV are less stringent and do not define JAK2-negative PV separately. In contrast, the BSH criteria are more stringent regarding hematocrit requirements and other additional criteria, including splenomegaly and white blood cell counts ([Table 2]). Although no guidelines recommend cytogenetic or molecular testing in cases with JAK2-negative erythrocytosis, the diagnostic workup of such cases must include the evaluation of both hereditary and acquired causes of erythrocytosis.[25] [26] Algorithm of workup of cases with polycythemia is shown in [Fig. 1].
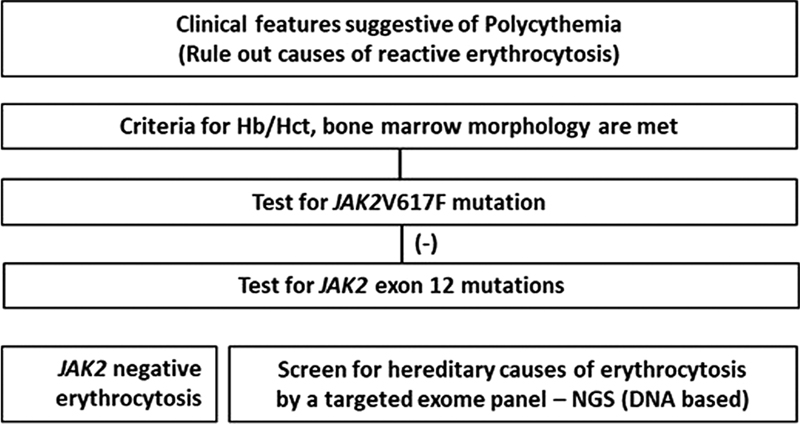
| Fig 1 : Algorithm for molecular testing in PV.
|
A1 |
Raised red cell mass (>25%-above predicted) or Hematocrit ≥ 0.60 in men & ≥ 0.56 in women |
|
A2 |
Absence of mutation in JAK2 |
|
A3 |
No cause of secondary erythrocytosis |
|
A4 |
Bone marrow histology consistent with polycythemia vera |
|
A5 |
Palpable splenomegaly |
|
A6 |
Presence of an acquired genetic abnormality (excluding BCR-ABL1) in the hematopoietic cells |
|
B1 |
Thrombocytosis (platelet count > 450 × 109/L) |
|
B2 |
Neutrophil leukocytosis (neutrophil count > 10 × 109/L in nonsmokers, ≥ 12.5 × 109/L in smokers) |
|
B3 |
Radiological evidence of splenomegaly |
|
B4 |
Low serum erythropoietin |
Essential Thrombocytosis
ET is a chronic MPN that primarily involves the megakaryocytes. In addition to sustained thrombocytosis (platelet count ≥ 450 × 109/L) in the peripheral blood, the disease is characterized by increased megakaryocyte counts in the bone marrow and thrombosis and bleeding.
In ET, approximately 55%-have JAK2V617F mutation, 5 to 10%-have MPL (exon 10) mutations, 20 to 25%-have CALR exon 9 mutations, and approximately 20%- are triple negative.[4] [5] [7] [8] [9] The absence of genetic or biological markers specific to ET makes it necessary to exclude other causes of thrombocytosis, including MPNs, inflammatory and infectious disorders, hemorrhage, other hematopoietic and non-hematopoietic neoplasms ([Supplementary Table S1], available in the online version).
Studies reporting noncanonical mutations in JAK2 and MPL genes in triple-negative ET warrant further investigation of these genes (not necessarily hotspot regions) and other genes associated with germline causes of thrombocytosis.[10] [11]
Primary Myelofibrosis
PMF is a clonal MPN characterized by an abnormal proliferation of megakaryocytes and granulocytes in the bone marrow, associated with fibrous connective tissue deposition and extramedullary hematopoiesis in fully developed disease. PMF is further subclassified as prefibrotic and overtly fibrotic PMF. Furthermore, approximately 15%-of patients with ET or PV might progress into a PMF-like phenotype (post-ET/PV MF) during their clinical course[3] ([Supplementary Table S2], available in the online version).
Molecular investigations in a patient with PMF include screening for driver mutations in the JAK2, CALR, and MPL genes, followed by mutations in other myeloid genes ([Table 3]). The algorithm of testing is shown in [Fig. 2].

| Figure 2:Algorithm for molecular testing in ET/PMF/CEL/CNL/MPN, NOS.
|
Gene |
Mutation location |
Mutation effect |
|---|---|---|
|
Phenotype driver mutations |
||
|
JAK2 |
Exon 14 |
JAK/STAT signaling |
|
JAK2 |
Exon 12 |
JAK/STAT signaling |
|
MPL |
Exon 10 |
JAK/STAT signaling |
|
CALR |
Exon 9 |
JAK/STAT signaling |
|
Initiation mutations |
||
|
TET2 |
All coding regions |
Epigenetic regulation |
|
DNMT3A |
Exons 7–23 |
Epigenetic regulation |
|
Disease-modifying mutations |
||
|
IDH1/2 |
Exon 4/ Exon 4 |
Epigenetic regulation |
|
EZH2 |
All coding regions |
Epigenetic regulation |
|
ASXL1 |
Exon 13 |
Epigenetic regulation |
|
SF3B1 |
Exon 12–16 |
mRNA splicing |
|
SRSF2 |
Exon 1 |
mRNA splicing |
|
U2AF1 |
Exons 2–7 |
mRNA splicing |
|
ZRSR2 |
All coding regions |
mRNA splicing |
|
RUNX1 |
All coding regions |
Transcription |
|
IKZF1 |
All coding regions |
Transcription |
|
TP53 |
All coding regions |
DNA repair |
|
CUX1 |
All coding regions |
Cell cycle |
|
CBL |
Exons 8–9 |
JAK/STAT signaling |
|
SH2B3 |
Exon 2 |
JAK/STAT signaling |
Detection of molecular markers is necessary to diagnose PMF and helps in these patients' risk stratification. The two new prognostic systems incorporating molecular markers in the risk stratification of PMF are GIPSS (genetically inspired prognostic scoring system) and MIPSS70+ version 2.0 (MIPSSv2; mutation- and karyotype-enhanced international prognostic scoring system).[27] [28]
The WHO 2016 recommends further screening of mutations in ASXL1, EZH2, TET2, IDH1, IDH2, SRSF2, and SF3B1 in cases of triple-negative PMF to establish clonality.[3] This heterogeneous group includes various somatic and germline mutations in driver genes and other myeloid neoplasia-associated genes.[10] [29] In a study where a larger panel of genes (where all coding regions of clinically relevant genes) was screened in a suspected case of pediatric myelofibrosis, a mutation in the MPIG6B gene (Megakaryocyte and platelet inhibitory receptor G6B- associated with thrombocytopenia, anemia, and myelofibrosis) was detected.[30] Screening for mutations in genes associated with germline causes of myelofibrosis is suggested in rare cases where the etiology of myelofibrosis remains elusive.
Chronic Neutrophilic Leukemia
CNL is a rare MPN. It is characterized by sustained peripheral blood neutrophilia, hypercellular bone marrow due to neutrophilic granulocyte proliferation, and hepatosplenomegaly. The diagnosis requires the absence of BCR::ABL1 fusion, excluding reactive neutrophilia and other myeloproliferative and myelodysplastic/MPNs ([Supplementary Table S3], available in the online version). CSF3R mutations are strongly, but not exclusively, associated with CNL and are a central diagnostic feature of this disorder.[3] Molecular investigations include screening by a targeted myeloid gene panel that helps establish a diagnosis (presence of CSF3R mutation) and identify adverse risk markers such as ASXL1 and SETBP1. Sequential FISH analysis or a targeted fusion gene panel screening by next-generation sequencing (NGS) (RNA-based), rearrangements in PDGFRA, PDGFRB, FGFR1, PCM1::JAK2, or other tyrosine kinase fusions must be excluded.
Chronic Eosinophilic Leukemia
CEL is a multisystem disease characterized by persistent hypereosinophilia and clonal proliferation of eosinophil precursors. It is clinically essential to distinguish between CEL and idiopathic hypereosinophilic syndrome (HES) clearly. The WHO defines idiopathic HES as eosinophilia (eosinophil count ≥ 1.5 × 109/L) persisting for more than or equal to 6 months (updated to 4 weeks in the WHO 2022) for which no underlying cause can be found, associated with signs of organ involvement and dysfunction and there is no evidence of eosinophil clonality.[2] [3]
The diagnosis of CEL is based on the exclusion of BCR::ABL1 fusion; rearrangement of PDGFRA, PDGFRB, or FGFR1; or PCM1::JAK2, ETV6::JAK2, or BCR::JAK2 fusions, ([Supplementary Table S4], available in the online version) which can be screened by either a sequential FISH analysis or a targeted fusion gene panel (RNA based). Extensive screening for mutations in genes associated with eosinophilia is suggested after all causes of reactive/ secondary eosinophilia are excluded.
Juvenile Myelomonocytic Leukemia
JMML is a hematopoietic stem cell-derived MPN of early childhood. Mechanisms involved in the pathogenesis in at least 90% of cases include unchecked activation of the RAS pathway. JMML diagnoses are made by clinical, laboratory, and molecular criteria ([Supplementary Table S5], available in the online version). There are three major changes to the diagnostic criteria in WHO 2022: (1) exclusion of KMT2a rearrangements; (2) removing monosomy 7 as a cytogenetic criterion; and (3) emphasizing the importance of diagnostic molecular studies, especially those that demonstrate the activation of the RAS pathway.[2] [3]
It is crucial to consider genetic factors in determining risk stratification, and treatment approaches for JMML, with cases originating from somatic mutations in PTPN11 and germline variants linked to neurofibromatosis type 1 (NF1) being the most aggressive. In contrast, spontaneous remission is seen in some cases with pathogenic germline CBL variants. Molecular investigations include screening for gene mutations associated with the RAS pathway by a targeted myeloid gene panel. When a mutation is identified in PTPN11, NF1, or CBL genes, the extended investigation must include establishing a germline/somatic etiology by testing DNA from the patient's skin fibroblasts for the same mutation. Once a germline etiology is established, family screening is suggested to identify the relatives at risk/related stem cell donors.
Myeloproliferative Neoplasm, Not Otherwise Specified
MPN (NOS) exhibits definite clinical, laboratory, molecular, and morphological characteristics of a MPN. However, it does not meet the diagnostic criteria for a specific MPN entity or overlaps the characteristics of two or more MPN entities ([Supplementary Table S6]).
Most cases of MPN, NOS fall into one of three groups:
A subset of cases with a masked prepolycythemic presentation of polycythemia vera, prefibrotic/early PMF, or early-phase essential thrombocythemia in which the characteristic features are not yet fully developed and a proportion of cases presenting with a portal or splanchnic vein thrombosis that fail to meet the diagnostic criteria for any of the specific MPN entities may also be considered to belong in this group.
MPN at an advanced stage, in which the underlying disorder is obscured by myelofibrosis, osteosclerosis, or a transformation to a more aggressive stage with increased blast counts and myelodysplastic changes.
Cases with convincing evidence of MPN but masked clinically and morphologically by a coexisting disorder.
Testing Methods for Driver Mutations and “Other/Passenger” Mutations in MPN
The molecular testing algorithm in MPN begins with the exclusion of BCR::ABL1 fusion. However, this is not recommended in cases with PV. Detection of BCR::ABL1 can be done either by conventional karyotyping/ FISH or reverse transcriptase polymerase chain reaction (PCR). Molecular screening investigations for driver and other mutations are usually performed on peripheral blood or bone marrow DNA.
Driver Mutations
JAK2
To detect JAK2V617F, there is a wide range of techniques available. Allele-specific PCR (AS PCR) is both sensitive and a specific test with a detection limit of 0.1%.[5] Other techniques include sanger or pyro sequencing, real-time/ quantitative allele-specific PCR, and NGS. Technique selection is based on several attributes, including a low threshold of detection (at least 1%-for diagnosis and 0.1%-for residual disease monitoring), a high level of specificity (no false positives at minimal detection levels), and a high degree of reproducibility. In cases of JAK2-negative polycythemia, mutations in JAK2 exon 12 mutations can be detected by sanger sequencing (sensitivity of 15–20%) or by NGS.[6]
CALR
There are three types of CALR mutations in MPN: insertions, deletions, or insertions and deletions (indels) in exon 9, which cause a +1 base pair frameshift of the coding sequence. This results in the generation of a translated protein lacking a retrieval sequence typical of normal CALR and endoplasmic reticulum resident proteins. CALR exon 9 mutations are categorized predominantly into a 52-bp deletion: (CALRdel52/type I; c.1092_1143del; L367fs*46) or a 5bp insertion CALRins5/type II; c1154_1155insTTGTC; K385fs*47). Other types include more than 50 different variations of CALR.[31] Because of the diversity and complexity of mutations that affect CALR exon 9, methodologies that can accommodate these mutations are most useful in the diagnostic setting. CALR mutation status is determined by PCR and capillary electrophoresis (fragment analysis and Sanger sequencing) of exon 9.[32]
MPL
Mutations in the MPL gene cluster around amino acid 515, which is in a stretch of five amino acids, found in the cytoplasmic section of the trans-membrane domain. W515L and W515K are the most common pathogenic mutations, while W515A, W515R, and W515S are rare mutations.[7] There are various detection methods available, but a multiplexed AS-PCR test that detects the four most common MPL exon 10 mutations (W515L, W515K, W515A, and S505N) with 100%- specificity and a 1%-sensitivity is the most cost-effective and currently being followed.[33]
While a multiplex AS PCR and fragment analysis are sensitive and specific, only the target mutations can be screened. However, a more cost-effective method is to screen a targeted panel of genes by NGS that includes all the coding regions of the driver genes along with other myeloid-associated genes, as it can detect both canonical and non-canonical mutations.[10] [11] Conventional methods of mutation detection are represented in [Fig. 3].
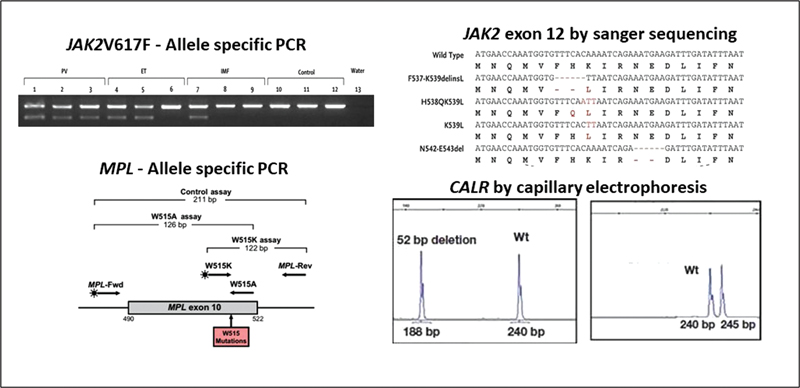
| Figure 3:Conventional methods of detecting JAK2V617F, MPL exon 10, and CALR exon 9 mutations.
Other (Passenger/Nondriver) MPN-Associated Mutations
Patients with MPNs have a highly complex genomic landscape with mutations in driver and nondriver genes conferring increased risk of disease progression and transformation to AML. As described earlier, somatic mutations in nondriver genes are frequently mutated in all MPN categories. Among these, mutations in the epigenetic regulator genes TET2, ASXL1, DNMT3A, and EZH2 and in the RNA splicing genes SRSF2 and U2AF1 are the commonest. Identification of mutations in these genes helps in the risk stratification in MPN patients. In cases where mutations in all the three driver genes are absent, further screening for mutations in ASXL1, EZH2, IDH1/2, SRSF2, TET2, and SF3B1 for evidence of clonality of hematopoiesis (triple-negative MPN) helps in differentiating clonal from reactive etiology. In nonclassical MPN, the exclusion of rearrangements in PDGFRA, PDGFRB, FGFR1, JAK2, or other TKI fusions is essential to make a diagnosis. These must be screened either by sequential FISH analysis or a targeted fusion gene panel. Myeloid-associated genes included in most targeted panels are represented in ([Table 3]).[34]
Discussion and Conclusion
With the ability to sequence multiple genes simultaneously and identify mutations within these genes with high specificity and sensitivity, NGS is ideal for the molecular diagnosis of MPNs.[32] Single-target assays may be employed sequentially, but multiplex assays, typically using NGS, sequence several targets in parallel and are more cost-effective. Either approach is acceptable if laboratory turnaround times and assay sensitivity are satisfactory [detection of 1–3%-variant allele frequency (VAF) or lower for JAK2 c.1849G > T (p.Val617Phe), and 5%-VAF for JAK2 exon 12, CALR exon nine or MPL exon ten variants]. The British Society for Hematology recommends that the use of broad myeloid NGS panels to screen cases with suspected MPN is unlikely to be cost-effective.[35] Still, if larger panels are used, the initial analysis and report should be limited to common MPN driver mutations.
The discovery of mutations in the three driver genes (JAK2, CALR, and MPL) has significantly contributed to understanding the classical BCR::ABL1-negative MPN. The introduction of mutations in one of the driver genes as a major WHO criterion highlights the importance of testing for these markers in the diagnosis, classification, and prognosis of various MPN subtypes. While the diagnosis of MPN remains multidisciplinary, requiring consideration of clinical features, histomorphological appearances, and other laboratory parameters, molecular diagnostics has become, and will continue to be, an increasingly integral part of this process.
Conflict of Interest
None declared.
Authors' Contributions
All the authors approve the submitted version of the manuscript. M.M. and A.K.A. wrote the paper. A.B.K., U.P.K., and B.P. contributed to the critical revisions of the manuscript.
Ethical Disclosure
None.
Supplementary Material
References
- 1 Pizzi M, Croci GA, Ruggeri M. et al. The classification of myeloproliferative neoplasms: rationale, historical background and future perspectives with focus on unclassifiable cases. Cancers (Basel) 2021; 13 (22) 5666
- 2 Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization classification of Haematolymphoid Tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36 (07) 1703-1719
- 3 Arber DA, Orazi A, Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127 (20) 2391-2405
- 4 Grinfeld J, Nangalia J, Baxter EJ. et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med 2018; 379 (15) 1416-1430
- 5 Baxter EJ, Scott LM, Campbell PJ. et al; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365 (9464): 1054-1061
- 6 Maddali M, Kulkarni UP, Ravindra N. et al. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Ann Hematol 2020; 99 (05) 983-989
- 7 Pardanani AD, Levine RL, Lasho T. et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108 (10) 3472-3476
- 8 Schnittger S, Bacher U, Eder C. et al. Molecular analyses of 15,542 patients with suspected BCR-ABL1-negative myeloproliferative disorders allow to develop a stepwise diagnostic workflow. Haematologica 2012; 97 (10) 1582-1585
- 9 Tefferi A. Myeloproliferative neoplasms: a decade of discoveries and treatment advances. Am J Hematol 2016; 91 (01) 50-58
- 10 Milosevic Feenstra JD, Nivarthi H, Gisslinger H. et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016; 127 (03) 325-332
- 11 Cabagnols X, Favale F, Pasquier F. et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016; 127 (03) 333-342
- 12 Acha P, Xandri M, Fuster-Tormo F. et al. Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am J Hematol 2019; 94 (10) E264-E267
- 13 Grinfeld J, Nangalia J, Green AR. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 2017; 102 (01) 7-17
- 14 Skoda RC, Duek A, Grisouard J. Pathogenesis of myeloproliferative neoplasms. Exp Hematol 2015; 43 (08) 599-608
- 15 Patel U, Luthra R, Medeiros LJ, Patel KP. Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin Lymphoma Myeloma Leuk 2017; 17S: S62-S74
- 16 Ernst T, Chase AJ, Score J. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42 (08) 722-726
- 17 Shirai CL, Ley JN, White BS. et al. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 2015; 27 (05) 631-643
- 18 Alduaij W, McNamara CJ, Schuh A. et al. Clinical utility of next-generation sequencing in the management of myeloproliferative neoplasms: a single-center experience. HemaSphere 2018; 2 (03) e44
- 19 Rotunno G, Pacilli A, Artusi V. et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: a study on 359 patients of the AGIMM group. Am J Hematol 2016; 91 (07) 681-686
- 20 Agarwal R, Blombery P, McBean M. et al. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: data from targeted next-generation sequencing in the diagnostic laboratory. Ann Hematol 2017; 96 (05) 725-732
- 21 Swerdlow SH, Campo E, Pileri SA. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127 (20) 2375-2390
- 22 Gerds AT, Gotlib J, Ali H. et al. Myeloproliferative neoplasms, version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2022; 20 (09) 1033-1062
- 23 Nann D, Fend F. Synoptic diagnostics of myeloproliferative neoplasms: morphology and molecular genetics. Cancers (Basel) 2021; 13 (14) 3528
- 24 Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 2020; 95 (12) 1599-1613
- 25 McMullin MF, Harrison CN, Ali S. et al; BSH Committee. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol 2019; 184 (02) 176-191
- 26 Cross NCP, Godfrey AL, Cargo C, Garg M, Mead AJ. A British Society for Haematology Good Practice Paper. The use of genetic tests to diagnose and manage patients with myeloproliferative and myeloproliferative/myelodysplastic neoplasms, and related disorders. Br J Haematol 2021; 195 (03) 338-351
- 27 Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 2021; 96 (01) 145-162
- 28 Guglielmelli P, Pietra D, Pane F. et al. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders-a consensus project of the Italian Society of Hematology. Leuk Res 2017; 58: 63-72
- 29 McClure RF, Ewalt MD, Crow J. et al. Clinical significance of DNA variants in chronic myeloid neoplasms: a report of the Association for molecular pathology. J Mol Diagn 2018; 20 (06) 717-737
- 30 Maddali M, Venkatraman A, Kulkarni UP. et al. Molecular characterization of triple-negative myeloproliferative neoplasms by next-generation sequencing. Ann Hematol 2022; 101 (09) 1987-2000
- 31 Klampfl T, Gisslinger H, Harutyunyan AS. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369 (25) 2379-2390
- 32 Langabeer SE, Andrikovics H, Asp J. et al; MPN&MPNr-EuroNet. Molecular diagnostics of myeloproliferative neoplasms. Eur J Haematol 2015; 95 (04) 270-279
- 33 Arunachalam AK, Suresh H, Mathews V, Balasubramanian P. Allele-specific PCR: a cost-effective screening method for MPL mutations in myeloproliferative neoplasms. Indian J Hematol Blood Transfus 2018; 34 (04) 765-767
- 34 Zoi K, Cross NCP. Genomics of myeloproliferative neoplasms. J Clin Oncol 2017; 35 (09) 947-954
- 35 Bench AJ, White HE, Foroni L. et al; British Committee for Standards in Haematology. Molecular diagnosis of the myeloproliferative neoplasms: UK guidelines for the detection of JAK2 V617F and other relevant mutations. Br J Haematol 2013; 160 (01) 25-34
Address for correspondence
Publication History
Article published online:
17 April 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig 1 : Algorithm for molecular testing in PV.
| Figure 2:Algorithm for molecular testing in ET/PMF/CEL/CNL/MPN, NOS.
| Figure 3:Conventional methods of detecting JAK2V617F, MPL exon 10, and CALR exon 9 mutations.
References
- 1 Pizzi M, Croci GA, Ruggeri M. et al. The classification of myeloproliferative neoplasms: rationale, historical background and future perspectives with focus on unclassifiable cases. Cancers (Basel) 2021; 13 (22) 5666
- 2 Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization classification of Haematolymphoid Tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36 (07) 1703-1719
- 3 Arber DA, Orazi A, Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127 (20) 2391-2405
- 4 Grinfeld J, Nangalia J, Baxter EJ. et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med 2018; 379 (15) 1416-1430
- 5 Baxter EJ, Scott LM, Campbell PJ. et al; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365 (9464): 1054-1061
- 6 Maddali M, Kulkarni UP, Ravindra N. et al. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Ann Hematol 2020; 99 (05) 983-989
- 7 Pardanani AD, Levine RL, Lasho T. et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108 (10) 3472-3476
- 8 Schnittger S, Bacher U, Eder C. et al. Molecular analyses of 15,542 patients with suspected BCR-ABL1-negative myeloproliferative disorders allow to develop a stepwise diagnostic workflow. Haematologica 2012; 97 (10) 1582-1585
- 9 Tefferi A. Myeloproliferative neoplasms: a decade of discoveries and treatment advances. Am J Hematol 2016; 91 (01) 50-58
- 10 Milosevic Feenstra JD, Nivarthi H, Gisslinger H. et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016; 127 (03) 325-332
- 11 Cabagnols X, Favale F, Pasquier F. et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016; 127 (03) 333-342
- 12 Acha P, Xandri M, Fuster-Tormo F. et al. Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am J Hematol 2019; 94 (10) E264-E267
- 13 Grinfeld J, Nangalia J, Green AR. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 2017; 102 (01) 7-17
- 14 Skoda RC, Duek A, Grisouard J. Pathogenesis of myeloproliferative neoplasms. Exp Hematol 2015; 43 (08) 599-608
- 15 Patel U, Luthra R, Medeiros LJ, Patel KP. Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin Lymphoma Myeloma Leuk 2017; 17S: S62-S74
- 16 Ernst T, Chase AJ, Score J. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42 (08) 722-726
- 17 Shirai CL, Ley JN, White BS. et al. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 2015; 27 (05) 631-643
- 18 Alduaij W, McNamara CJ, Schuh A. et al. Clinical utility of next-generation sequencing in the management of myeloproliferative neoplasms: a single-center experience. HemaSphere 2018; 2 (03) e44
- 19 Rotunno G, Pacilli A, Artusi V. et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: a study on 359 patients of the AGIMM group. Am J Hematol 2016; 91 (07) 681-686
- 20 Agarwal R, Blombery P, McBean M. et al. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: data from targeted next-generation sequencing in the diagnostic laboratory. Ann Hematol 2017; 96 (05) 725-732
- 21 Swerdlow SH, Campo E, Pileri SA. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127 (20) 2375-2390
- 22 Gerds AT, Gotlib J, Ali H. et al. Myeloproliferative neoplasms, version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2022; 20 (09) 1033-1062
- 23 Nann D, Fend F. Synoptic diagnostics of myeloproliferative neoplasms: morphology and molecular genetics. Cancers (Basel) 2021; 13 (14) 3528
- 24 Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 2020; 95 (12) 1599-1613
- 25 McMullin MF, Harrison CN, Ali S. et al; BSH Committee. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol 2019; 184 (02) 176-191
- 26 Cross NCP, Godfrey AL, Cargo C, Garg M, Mead AJ. A British Society for Haematology Good Practice Paper. The use of genetic tests to diagnose and manage patients with myeloproliferative and myeloproliferative/myelodysplastic neoplasms, and related disorders. Br J Haematol 2021; 195 (03) 338-351
- 27 Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 2021; 96 (01) 145-162
- 28 Guglielmelli P, Pietra D, Pane F. et al. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders-a consensus project of the Italian Society of Hematology. Leuk Res 2017; 58: 63-72
- 29 McClure RF, Ewalt MD, Crow J. et al. Clinical significance of DNA variants in chronic myeloid neoplasms: a report of the Association for molecular pathology. J Mol Diagn 2018; 20 (06) 717-737
- 30 Maddali M, Venkatraman A, Kulkarni UP. et al. Molecular characterization of triple-negative myeloproliferative neoplasms by next-generation sequencing. Ann Hematol 2022; 101 (09) 1987-2000
- 31 Klampfl T, Gisslinger H, Harutyunyan AS. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369 (25) 2379-2390
- 32 Langabeer SE, Andrikovics H, Asp J. et al; MPN&MPNr-EuroNet. Molecular diagnostics of myeloproliferative neoplasms. Eur J Haematol 2015; 95 (04) 270-279
- 33 Arunachalam AK, Suresh H, Mathews V, Balasubramanian P. Allele-specific PCR: a cost-effective screening method for MPL mutations in myeloproliferative neoplasms. Indian J Hematol Blood Transfus 2018; 34 (04) 765-767
- 34 Zoi K, Cross NCP. Genomics of myeloproliferative neoplasms. J Clin Oncol 2017; 35 (09) 947-954
- 35 Bench AJ, White HE, Foroni L. et al; British Committee for Standards in Haematology. Molecular diagnosis of the myeloproliferative neoplasms: UK guidelines for the detection of JAK2 V617F and other relevant mutations. Br J Haematol 2013; 160 (01) 25-34