 PDF
PDF  Views
Views  Share
Share


A Case of Synchronous Malignancy with Novel Missense Mutation in a Child: Is This Li-Fraumeni Syndrome or a Novel Case Masquerading as Li-Fraumeni Syndrome
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(05): 777-780
DOI: DOI: 10.4103/ijmpo.ijmpo_202_20
Abstract
We report the case of an 11-month-old child who presented with a change in voice, increased weight, and hirsutism, who was also found to have elevated levels of serum cortisol and testosterone, showing three synchronous malignancies in the liver, left adrenal gland, and posterior mediastinum. Clinical exome sequencing report revealed germline TP53 (P177A) and MLH3 (V741P) mutations with NMYC positive neuroblastoma. At the outset, this may look like a Li-Fraumeni syndrome (LFS) with TP53 germline mutation but lacks other features to be termed as LFS or Li-Fraumeni-like syndrome. The gene mutation variant found in this case (P177A) is a novel missense mutation which has never been reported, and the MLH3 gene mutation variant V741P has not been previously associated with any of the malignancies seen in this child.
Keywords
Publication History
Received: 27 April 2020
Accepted: 23 August 2020
Article published online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
We report the case of an 11-month-old child who presented with a change in voice, increased weight, and hirsutism, who was also found to have elevated levels of serum cortisol and testosterone, showing three synchronous malignancies in the liver, left adrenal gland, and posterior mediastinum. Clinical exome sequencing report revealed germline TP53 (P177A) and MLH3 (V741P) mutations with NMYC positive neuroblastoma. At the outset, this may look like a Li-Fraumeni syndrome (LFS) with TP53 germline mutation but lacks other features to be termed as LFS or Li-Fraumeni-like syndrome. The gene mutation variant found in this case (P177A) is a novel missense mutation which has never been reported, and the MLH3 gene mutation variant V741P has not been previously associated with any of the malignancies seen in this child.
Keywords
Adrenocortical malignancy - hepatoblastoma - mismatch repair gene - neuroblastoma - synchronous malignancyIntroduction
Synchronous malignancies are rare occurrences in children and pose a challenge in terms of chemotherapy and surgery.[1] This challenge is intensified if there is an association with TP53 which predisposes the individual to development of tumors when exposed to chemotherapy or radiotherapy.[2] This is such a case which had high-risk malignancies otherwise treated by high-dose chemotherapy and/or radiotherapy, but because associated with TP53 and MLH3 gene mutations, management strategy had to be changed. The rarity of the gene mutations associated makes it a unique case. Li-Fraumeni syndrome (LFS) is a familial cancer syndrome associated with multiple malignancies and TP53 gene mutation.[3] This case report comes close to being labeled as LFS.
Case Report
An 11-month-old male child was admitted with the complaints of hoarseness of voice and hirsutism from 8 to 9 months of age. The child had generalized coarse hair overgrowth over the entire body along with weight gain for which an endocrine consultation was done. Hormonal assay and radiological imaging studies were done after the endocrine consult.
Medical investigations revealed high levels of serum cortisol (20.85 μg/dL) and elevated testosterone levels (free testosterone 250 ng/dL and dihydrotestosterone 2500 ng/dL). Urinary vanillylmandelic acid levels were normal, and alpha-fetoprotein level raised to 912 ng/mL (normal < 10 ng/mL).
Radiological investigation revealed a mass in the left adrenal gland, which was heterogeneous and well encapsulated with areas of focal calcification seen within the tumor [Figure 1a]. There was an exophytic mass in the segment VI of the liver seen without any extrahepatic, inferior vena cava (IVC), portal vein, or nodal involvement [Figure 1b]. Computed tomography also revealed a well-encapsulated mass in the posterior mediastinum extending between the T4 and T7 vertebrae without any intraspinal extensions [Figure 1c]. The lungs were clear of metastasis, so was the brain.
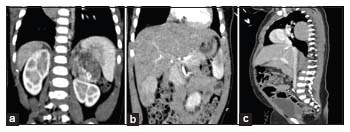
| Figure 1: (a) Radiological image of adrenocortical carcinoma, (b) radiological image of hepatoblastoma, (c) radiological image of neuroblastoma
There were no dysmorphic features present in the child and no history of malignancy in the siblings.
The mother had a history of six previous abortions and hormonal treatment during pregnancy. The abortions were not spontaneous but for wanting a male child. The child's father was formerly treated for renal cell carcinoma, underwent partial nephrectomy 5 years ago, has not received any chemotherapy postprocedure, and is symptom free. There was no other family history of any malignancy.
There were three masses in this child and in order to get a bearing as to which is primary and which is metastasis, we decided to biopsy one of them and the safest was to perform a liver biopsy. He underwent an ultrasound-guided liver biopsy under corticosteroid cover, and it was reported as fetal variant hepatoblastoma (ICD-O-3 code 8970). Further, a right thoracoscopic biopsy was done to rule out metastasis, but it was reported to be a neuroblastoma stroma poor type (ICD-O-3 code 9490).
After ruling out adrenal medullary involvement (by urinary vanillylmandelic acid and homovanillic acid level) to prevent the intraoperative adrenal storm, he underwent a left adrenalectomy [Figure 2a] and excision of hepatoblastoma under corticosteroid cover [Figure 2b]. The left adrenal mass was reported as adrenocortical malignancy (ICD-O-3 code 8370) without a breach in the capsule and the hepatoblastoma was of fetal variant without any hepatic vein, IVC, or extrahepatic involvement and clear margins of resection. The chest mass was excised 2 weeks later, and its biopsy was reported as neuroblastoma, which was well differentiated and of stroma poor type [Figure 2c].
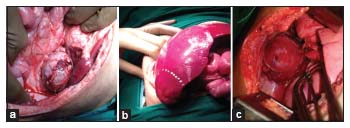
| Figure 2: (a) Intraoperative picture of adrenocortical carcinoma, (b) intraoperative picture of hepatoblastoma, (c) intraoperative picture of neuroblastoma
Hormonal evaluation post tumor excision revealed a reduction in the levels of cortisol (2.0 μg/dL), testosterone (free testosterone 0.1 ng/dL and dihydrotestosterone 27 ng/dL), and alpha-fetoprotein (3.0 ng/mL). Currently, the levels of the elevated hormones have normalized, and he is off steroids which were stopped after tapering doses.
Because the child has three histologically distinct malignancies, gene mutation studies were done which revealed germline TP53 gene mutation (c. C530G, p. Pro177Arg) with allelic ID 468390 and MLH3 gene (c. G2221T, p. Val741Phe) mutations. Interphase fluorescent in situ hybridization (FISH) technique revealed a 15-fold increase of NMYC signals to chromosome enumeration probe signals, confirming NMYC amplification. Following this, multiplex ligation-dependent probe amplification (MLPA) assay was also performed on the neuroblastoma tissue to check for segmental chromosomal anomalies on chromosomes 1, 2, 3, 4, 7, 9, 11, 12, and 17 [Table 1]. The parents were offered to undergo genetic testing to find out any genetic abnormalities, but they refused to undergo such tests.
Table 1Genetic and molecular findings on multiplex ligation-dependent probe amplification and fluorescent in situ hybridization in cases of neuroblastoma and its significance
|
Genetic alteration |
Copy status |
Significance |
|---|---|---|
|
ALK |
Normal |
Helps activating NMYC, highrisk tumor |
|
DDX1 |
Amplification |
High-risk tumor |
|
NMYC |
Amplification |
High-risk tumor |
|
1p |
None |
Near-diploid or tetraploid tumors |
|
11q23 |
Normal |
Higher disease state and poor prognosis |
|
17q |
Gains |
Poor outcome |
|
2p |
Amplification |
Very aggressive course |
|
3p |
Normal |
Tumor suppressor gene, higher age at diagnosis |
|
4p |
Normal |
Tumor suppressor gene |
|
7q |
Normal |
Tumor suppressor gene |
Discussion
Primary multiple tumors are defined as malignancies with different histopathological origin in one person. Its increased incidence seen nowadays is owing to better diagnostic techniques and improved treatment methods.[4] Multiple malignancies are reported along with TP53 mutation and LFS. Two distinct histological types of malignancies predominate the TP53 mutation reports. The most commonly associated malignancies with germline TP53 mutations and LFS are the adrenocortical malignancies, soft-tissue sarcomas, brain tumors, and breast tumors.[5]
There are multiple criteria which have been laid down for the identification of LFS which are classic and Chompret.[6] For those who do not fit into the classic criteria, Birch and Eeles have laid down criteria for identifying Li-Fraumeni-like syndrome (LFL). There is another syndrome which is described in literature called hereditary neoplastic syndrome. This is also associated with multiple malignancies and characterized by earlier onset of tumors than their described age. However, these are associated with endocrine tumors.[7]
Our patient does not fit into the LFS or LFL with any of the criteria laid down even with the presence of a renal cell carcinoma in his father. This makes our case a unique one with germline TP53 mutation and three histologically different malignancies (one of them being adrenocortical carcinoma) not matching the criteria for LFS and having malignancies other than the ones usually reported with LFS or LFL. This is a novel mutation, but its clinical significance needs to be proven in future.
Considering this case as a novel case of synchronous malignancies, the age of presentation also makes this unique, as most commonly the multiple malignancy syndromes are seen in the second or third decade.[8] This makes our case a unique one as the three distinct histopathological malignancies are reported at the age of 11 months. Three other reports were found where the patients are aged 6, 8, and 10 months.[3],[9],[10]
There are multiple cases of neuroblastoma which are reported with TP53 gene mutation [Table 2]. There are six reported cases of TP53 mutation with adrenocortical cancer (ACC) and neuroblastoma. This case is seventh such report.
Table 2 Cases of neuroblastoma combined with other tumors and TP53 mutation
The reported mutation in TP53 gene in our present case is a missense mutation c. C530G, p. Pro177Arg (P177A) with allelic ID 468390. This germline mutation has been identified in scientific literature to be present within the DNA-binding domain of TP53 but has not been biochemically characterized, and, therefore, its effect on TP53 protein function is still unknown, although speculated to have a partially gain-of-function mutation.[13] According to the International Agency for Research on Cancer (IARC) TP53 database, there has been only one somatic TP53 P177A mutation identified in colorectal cancer and no germline TP53 P177A has been identified till August 2018. Totally, there are 498 gene variants identified with TP53 mutation in the IARC TP53 database.[14]
The clinical exome sequencing also identified a mutation in the MLH3 gene which was a missense mutation c. G2221T, pVal741Phe (V741P) which is associated with hereditary nonpolyposis colorectal cancer (HNPCC) type 7. This type of HNPCC is known to cause early-onset colorectal, endometrial, gastric, and breast cancers.[15] According to the Centre for Genomic Study and Genetics, Zhejiang University, there are a total of 54 gene variants which are described for MLH3 mutation and most of them are on exon 2, as in our case. According to the same genetic registry, there have been only two instances of reports with V741P mutation, which were seen in adult familial endometrial carcinoma and colorectal cancer.[16]
The neuroblastoma tissue which was subjected to check for NMYC amplification test by interphase FISH assay revealed a 15-fold increase in the NMYC signals. Further, MLPA assay was performed to identify segmental chromosomal anomalies, which revealed alterations as tabulated in [Table 1].
This case is particularly unique due to presentation with three histologically malignant tumors at 11 months of age. It did not fit into the criteria laid down for LFS in the presence of TP53 mutation and ACC with neuroblastoma. The gene mutation variant found is a novel missense mutation which has never been reported in germline mutation, while only one case is reported that of a somatic mutation (with colorectal cancer).[14] The MLH3 gene mutation variant V741P has not been previously associated with any of the malignancies seen in this child.[16] This indirectly may point toward the child developing any other malignancy association with this genetic aberration later in his life.
The MLPA assay and NMYC amplification rendered the neuroblastoma as a high-risk tumor. The chemotherapy regimen for high-risk neuroblastoma with NMYC, DDX1, and 2p amplifications consists of high-dose chemotherapy, irradiation, followed by immunotherapy.[17] The treating doctors were pushed against the wall for deciding the therapy as radiotherapy or high-dose chemotherapy cannot be performed because of TP53 mutation as these patients are known to develop more malignancies when exposed to radiation or other cytotoxic drugs.
A management protocol for these patients with multiple malignancies cannot be formulated as these are one-off presentation and each such presentation must be dealt individually.
Conclusion – Take-Home Message
Multiple synchronous malignancies are a rare occurrence in children. Cytogenetics (karyotyping, FISH, and comparative genomic hybridization) and molecular cytogenetics (MLPA and restriction fragment length polymorphism) in these cases help identifying the known mutations, whereas molecular cytogenetic techniques (denaturing gradient gel electrophoresis, single-strand conformational polymorphism, and heteroduplex analysis) will identify the unknown mutations. Genetic mapping of parents and siblings is crucial to identify inherited mutations or de novo mutations. Establishing a chemotherapy regimen and tailoring it for these patients requires a multidisciplinary approach. Follow-up of these children with an eye out for a possible malignancy is of utmost importance.
Consent
A written informed consent has been obtained from the parents of this patient for publication of this case report and accompanying images. A copy will be made available at request.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient's parents have given consent for their child's images and other clinical information to be reported in the journal. The patient's parents understand that their child's name and initial will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- Pivnick EK, Furman WL, Velagaleti GV, Jenkins JJ, Chase NA, Ribeiro RC. Simultaneous adrenocortical carcinoma and ganglioneuroblastoma in a child with Turner syndrome and germline p53 mutation. J Med Genet 1998; 35: 328-32
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D. et al. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013; 45: 279-84
- Tang YJ, Yu TT, Ma J, Zhou Y, Xu M, Gao YJ. Composite adrenocortical carcinoma and neuroblastoma in an infant with a TP53 germline mutation: A case report and literature review. J Pediatr Hematol Oncol 2019; 41: 399-401
- Moertel CG, Dockerty MB, Baggenstoss AH. Multiple primary malignant neoplasms. I. Introduction and presentation of data. Cancer 1961; 14: 221-30
- Behjati S, Maschietto M, Williams RD, Side L, Hubank M, West R. et al. A pathogenic mosaic TP53 mutation in two germ layers detected by next generation sequencing. PLoS One 2014; 9: e96531
- Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E. et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009; 27: e108-9
- Frank TS. Hereditary cancer syndromes. Arch Pathol Lab Med 2001; 125: 85-90
- Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ. et al. Beyond Li-Fraumeni syndrome: Clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27: 1250-6
- Courtney R, Ranganathan S. Simultaneous adrenocortical carcinoma and neuroblastoma in an infant with a novel germline p53 mutation. J Pediatr Hematol Oncol 2015; 37: 215-8
- Rossbach HC, Baschinsky D, Wynn T, Obzut D, Sutcliffe M, Tebbi C. Composite adrenal anaplastic neuroblastoma and virilizing adrenocortical tumor with germline TP53 R248W mutation. Pediatr Blood Cancer 2008; 50: 681-3
- Seidinger AL, Fortes FP, Mastellaro MJ, Cardinalli IA, Zambaldi LG, Aguiar SS. et al. Occurrence of neuroblastoma among TP53 p.R337H Carriers. PLoS One 2015; 10: e0140356
- Porter DE, Holden ST, Steel CM, Cohen BB, Wallace MR, Reid R. A significant proportion of patients with osteosarcoma may belong to Li-Fraumeni cancer families. J Bone Joint Surg Br 1992; 74: 883-6
- Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 2018; 71: 178-90, e8
- IARC TP53 Search. http://p53.iarc.fr/TP53GeneVariations.aspx. Available from: [Last accessed on 2019 Jul 18]
- Liu HX, Zhou XL, Liu T, Werelius B, Lindmark G, Dahl N. et al. The role of hMLH3 in familial colorectal cancer. Cancer Res 2003; 63: 1894-9
- All genes-Zhejiang University Center for Genetic and Genomic Medicine (ZJU-CGGM) & Dian Diagnostics. http://www.genomed.zju.edu.cn/LOVD3/gene. Available from: [Last accessed on 2019 Jul 18]
- Casey DL, Kushner BH, Cheung NK, Modak S, LaQuaglia MP. et al. Local control with 21-Gy radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 2016; 96: 393-400
Address for correspondence
Publication History
Received: 27 April 2020
Accepted: 23 August 2020
Article published online:
17 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: (a) Radiological image of adrenocortical carcinoma, (b) radiological image of hepatoblastoma, (c) radiological image of neuroblastoma
| Figure 2: (a) Intraoperative picture of adrenocortical carcinoma, (b) intraoperative picture of hepatoblastoma, (c) intraoperative picture of neuroblastoma
References
- Pivnick EK, Furman WL, Velagaleti GV, Jenkins JJ, Chase NA, Ribeiro RC. Simultaneous adrenocortical carcinoma and ganglioneuroblastoma in a child with Turner syndrome and germline p53 mutation. J Med Genet 1998; 35: 328-32
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D. et al. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013; 45: 279-84
- Tang YJ, Yu TT, Ma J, Zhou Y, Xu M, Gao YJ. Composite adrenocortical carcinoma and neuroblastoma in an infant with a TP53 germline mutation: A case report and literature review. J Pediatr Hematol Oncol 2019; 41: 399-401
- Moertel CG, Dockerty MB, Baggenstoss AH. Multiple primary malignant neoplasms. I. Introduction and presentation of data. Cancer 1961; 14: 221-30
- Behjati S, Maschietto M, Williams RD, Side L, Hubank M, West R. et al. A pathogenic mosaic TP53 mutation in two germ layers detected by next generation sequencing. PLoS One 2014; 9: e96531
- Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E. et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009; 27: e108-9
- Frank TS. Hereditary cancer syndromes. Arch Pathol Lab Med 2001; 125: 85-90
- Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ. et al. Beyond Li-Fraumeni syndrome: Clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27: 1250-6
- Courtney R, Ranganathan S. Simultaneous adrenocortical carcinoma and neuroblastoma in an infant with a novel germline p53 mutation. J Pediatr Hematol Oncol 2015; 37: 215-8
- Rossbach HC, Baschinsky D, Wynn T, Obzut D, Sutcliffe M, Tebbi C. Composite adrenal anaplastic neuroblastoma and virilizing adrenocortical tumor with germline TP53 R248W mutation. Pediatr Blood Cancer 2008; 50: 681-3
- Seidinger AL, Fortes FP, Mastellaro MJ, Cardinalli IA, Zambaldi LG, Aguiar SS. et al. Occurrence of neuroblastoma among TP53 p.R337H Carriers. PLoS One 2015; 10: e0140356
- Porter DE, Holden ST, Steel CM, Cohen BB, Wallace MR, Reid R. A significant proportion of patients with osteosarcoma may belong to Li-Fraumeni cancer families. J Bone Joint Surg Br 1992; 74: 883-6
- Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 2018; 71: 178-90, e8
- IARC TP53 Search. http://p53.iarc.fr/TP53GeneVariations.aspx. Available from: [Last accessed on 2019 Jul 18]
- Liu HX, Zhou XL, Liu T, Werelius B, Lindmark G, Dahl N. et al. The role of hMLH3 in familial colorectal cancer. Cancer Res 2003; 63: 1894-9
- All genes-Zhejiang University Center for Genetic and Genomic Medicine (ZJU-CGGM) & Dian Diagnostics. http://www.genomed.zju.edu.cn/LOVD3/gene. Available from: [Last accessed on 2019 Jul 18]
- Casey DL, Kushner BH, Cheung NK, Modak S, LaQuaglia MP. et al. Local control with 21-Gy radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 2016; 96: 393-400